

Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding
- PMID: 25551548
- PMCID: PMC4601269
- DOI: 10.4161/19336896.2014.983398
Exosome-dependent and independent mechanisms are involved in prion-like transmission of propagated Cu/Zn superoxide dismutase misfolding
Abstract
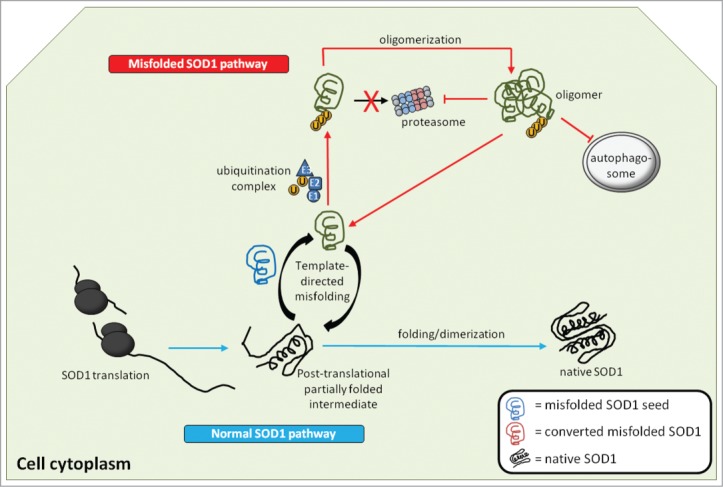
Amyotrophic lateral sclerosis (ALS), a fatal adult-onset degenerative neuromuscular disorder with a poorly defined etiology, progresses in an orderly spatiotemporal manner from one or more foci within the nervous system, reminiscent of prion disease pathology. We have previously shown that misfolded mutant Cu/Zn superoxide dismutase (SOD1), mutation of which is associated with a subset of ALS cases, can induce endogenous wild-type SOD1 misfolding in the intracellular environment in a templating fashion similar to that of misfolded prion protein. Our recent observations further extend the prion paradigm of pathological SOD1 to help explain the intercellular transmission of disease along the neuroaxis. It has been shown that both mutant and misfolded wild-type SOD1 can traverse cell-to-cell either as protein aggregates that are released from dying cells and taken up by neighboring cells via macropinocytosis, or released to the extracellular environment on the surface of exosomes secreted from living cells. Furthermore, once propagation of misfolded wild-type SOD1 has been initiated in human cell culture, it continues over multiple passages of transfer and cell growth. Propagation and transmission of misfolded wild-type SOD1 is therefore a potential mechanism in the systematic progression of ALS pathology.
Keywords: amyotrophic lateral sclerosis; exosome; intercellular transmission; prion-like; protein misfolding; superoxide dismutase.
Figures

Similar articles
-
Disease Mechanisms in ALS: Misfolded SOD1 Transferred Through Exosome-Dependent and Exosome-Independent Pathways.Cell Mol Neurobiol. 2016 Apr;36(3):377-81. doi: 10.1007/s10571-015-0294-3. Epub 2016 Feb 23. Cell Mol Neurobiol. 2016. PMID: 26908139 Review.
-
Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms.Proc Natl Acad Sci U S A. 2014 Mar 4;111(9):3620-5. doi: 10.1073/pnas.1312245111. Epub 2014 Feb 18. Proc Natl Acad Sci U S A. 2014. PMID: 24550511 Free PMC article.
-
Intercellular Prion-Like Conversion and Transmission of Cu/Zn Superoxide Dismutase (SOD1) in Cell Culture.Methods Mol Biol. 2017;1658:357-367. doi: 10.1007/978-1-4939-7244-9_24. Methods Mol Biol. 2017. PMID: 28861801
-
From molecule to molecule and cell to cell: prion-like mechanisms in amyotrophic lateral sclerosis.Neurobiol Dis. 2015 May;77:257-65. doi: 10.1016/j.nbd.2015.02.009. Epub 2015 Feb 17. Neurobiol Dis. 2015. PMID: 25701498 Review.
-
A Metal-Free, Disulfide Oxidized Form of Superoxide Dismutase 1 as a Primary Misfolded Species with Prion-Like Properties in the Extracellular Environments Surrounding Motor Neuron-Like Cells.Int J Mol Sci. 2021 Apr 16;22(8):4155. doi: 10.3390/ijms22084155. Int J Mol Sci. 2021. PMID: 33923808 Free PMC article.
Cited by
-
CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1.J Biol Chem. 2019 Mar 8;294(10):3744-3759. doi: 10.1074/jbc.RA118.004825. Epub 2019 Jan 11. J Biol Chem. 2019. PMID: 30635404 Free PMC article.
-
Cysteine String Protein Controls Two Routes of Export for Misfolded Huntingtin.Front Neurosci. 2022 Jan 5;15:762439. doi: 10.3389/fnins.2021.762439. eCollection 2021. Front Neurosci. 2022. PMID: 35069097 Free PMC article.
-
Exosomes in the Pathology of Neurodegenerative Diseases.J Biol Chem. 2016 Dec 23;291(52):26589-26597. doi: 10.1074/jbc.R116.757955. Epub 2016 Nov 16. J Biol Chem. 2016. PMID: 27852825 Free PMC article. Review.
-
Doxorubicin-Loaded Extracellular Vesicles Enhance Tumor Cell Death in Retinoblastoma.Bioengineering (Basel). 2022 Nov 10;9(11):671. doi: 10.3390/bioengineering9110671. Bioengineering (Basel). 2022. PMID: 36354582 Free PMC article.
-
Advances in Ferritin Physiology and Possible Implications in Bacterial Infection.Int J Mol Sci. 2023 Feb 28;24(5):4659. doi: 10.3390/ijms24054659. Int J Mol Sci. 2023. PMID: 36902088 Free PMC article. Review.
References
-
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001; 2:806-19; PMID:11715057; http://dx.doi.org/10.1038/35097565 - DOI - PubMed
-
- Strong M, Rosenfeld J. Amyotrophic lateral sclerosis: a review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord 2003; 4:136-43; PMID:13129799; http://dx.doi.org/10.1080/14660820310011250 - DOI - PubMed
-
- Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 1995; 118 (Pt 3):707-19; PMID:7600088; http://dx.doi.org/10.1093/brain/118.3.707 - DOI - PubMed
-
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013; 14:248-64; PMID:23463272; http://dx.doi.org/10.1038/nrn3430 - DOI - PubMed
-
- Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A, et al. . Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 2012; 123:409-17; PMID:22228244; http://dx.doi.org/10.1007/s00401-011-0937-5 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous