

INS-gene mutations: from genetics and beta cell biology to clinical disease
- PMID: 25542748
- PMCID: PMC4404187
- DOI: 10.1016/j.mam.2014.12.001
INS-gene mutations: from genetics and beta cell biology to clinical disease
Abstract
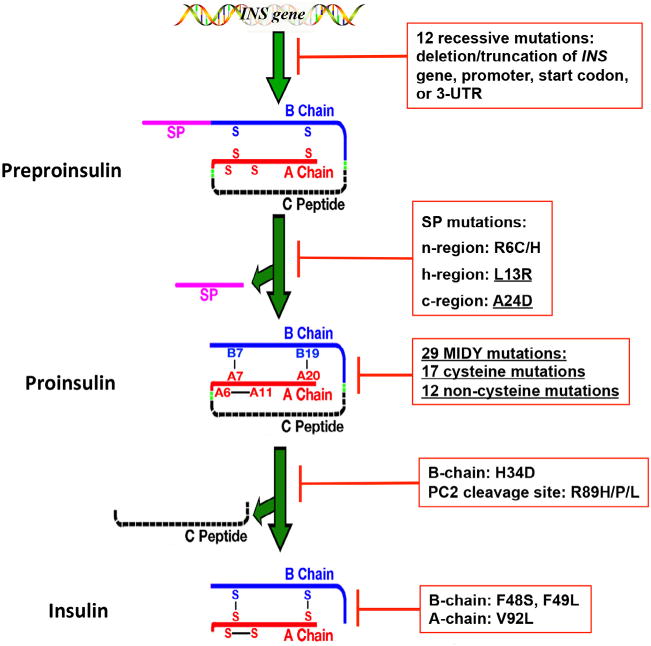
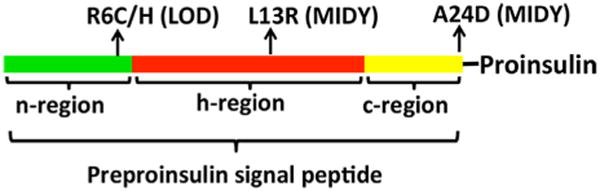
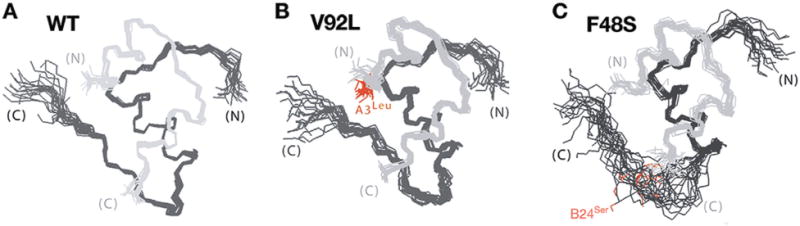
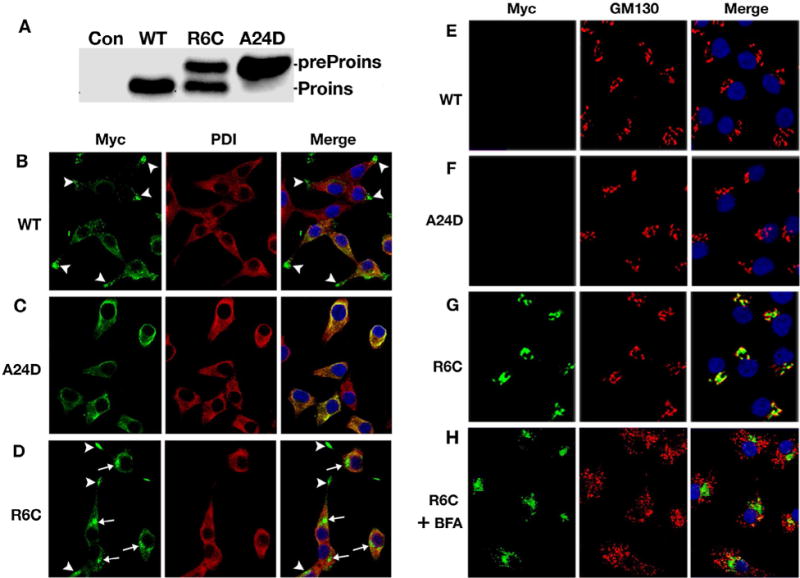
A growing list of insulin gene mutations causing a new form of monogenic diabetes has drawn increasing attention over the past seven years. The mutations have been identified in the untranslated regions of the insulin gene as well as the coding sequence of preproinsulin including within the signal peptide, insulin B-chain, C-peptide, insulin A-chain, and the proteolytic cleavage sites both for signal peptidase and the prohormone convertases. These mutations affect a variety of different steps of insulin biosynthesis in pancreatic beta cells. Importantly, although many of these mutations cause proinsulin misfolding with early onset autosomal dominant diabetes, some of the mutant alleles appear to engage different cellular and molecular mechanisms that underlie beta cell failure and diabetes. In this article, we review the most recent advances in the field and discuss challenges as well as potential strategies to prevent/delay the development and progression of autosomal dominant diabetes caused by INS-gene mutations. It is worth noting that although diabetes caused by INS gene mutations is rare, increasing evidence suggests that defects in the pathway of insulin biosynthesis may also be involved in the progression of more common types of diabetes. Collectively, the (pre)proinsulin mutants provide insightful molecular models to better understand the pathogenesis of all forms of diabetes in which preproinsulin processing defects, proinsulin misfolding, and ER stress are involved.
Keywords: Diabetes; Endoplasmic reticulum stress; Insulin biosynthesis; Insulin gene mutation; Pancreatic beta cell; Proinsulin misfolding.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells.Vitam Horm. 2014;95:35-62. doi: 10.1016/B978-0-12-800174-5.00002-8. Vitam Horm. 2014. PMID: 24559913 Review.
-
Biosynthesis, structure, and folding of the insulin precursor protein.Diabetes Obes Metab. 2018 Sep;20 Suppl 2(Suppl 2):28-50. doi: 10.1111/dom.13378. Diabetes Obes Metab. 2018. PMID: 30230185 Free PMC article. Review.
-
Biological behaviors of mutant proinsulin contribute to the phenotypic spectrum of diabetes associated with insulin gene mutations.Mol Cell Endocrinol. 2020 Dec 1;518:111025. doi: 10.1016/j.mce.2020.111025. Epub 2020 Sep 8. Mol Cell Endocrinol. 2020. PMID: 32916194 Free PMC article.
-
Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes.Ann N Y Acad Sci. 2018 Apr;1418(1):5-19. doi: 10.1111/nyas.13531. Epub 2018 Jan 28. Ann N Y Acad Sci. 2018. PMID: 29377149 Free PMC article. Review.
-
Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes.Mol Aspects Med. 2015 Apr;42:105-18. doi: 10.1016/j.mam.2015.01.001. Epub 2015 Jan 8. Mol Aspects Med. 2015. PMID: 25579745 Free PMC article. Review.
Cited by
-
Novel Approach for Treating Diabetes in a Patient With the Heterozygous Pathogenic Variant R46Q in the Insulin Gene.JCEM Case Rep. 2024 Jul 18;2(7):luae134. doi: 10.1210/jcemcr/luae134. eCollection 2024 Jul. JCEM Case Rep. 2024. PMID: 39027635 Free PMC article.
-
Two siblings with a rare type of maturity-onset diabetes of the young (MODY).BMJ Case Rep. 2023 Feb 10;16(2):e249362. doi: 10.1136/bcr-2022-249362. BMJ Case Rep. 2023. PMID: 36764736
-
Pharmacological Inhibition of Inositol-Requiring Enzyme 1α RNase Activity Protects Pancreatic Beta Cell and Improves Diabetic Condition in Insulin Mutation-Induced Diabetes.Front Endocrinol (Lausanne). 2021 Oct 5;12:749879. doi: 10.3389/fendo.2021.749879. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 34675883 Free PMC article.
-
Misfolded proinsulin impairs processing of precursor of insulin receptor and insulin signaling in β cells.FASEB J. 2019 Oct;33(10):11338-11348. doi: 10.1096/fj.201900442R. Epub 2019 Aug 1. FASEB J. 2019. PMID: 31311313 Free PMC article.
-
Predisposition to Proinsulin Misfolding as a Genetic Risk to Diet-Induced Diabetes.Diabetes. 2021 Nov;70(11):2580-2594. doi: 10.2337/db21-0422. Epub 2021 Aug 30. Diabetes. 2021. PMID: 34462258 Free PMC article.
References
-
- Alarcón C, Lincoln B, Rhodes CJ. The biosynthesis of the subtilisin-related proprotein convertase PC3, but no that of the PC2 convertase, is regulated by glucose in parallel to proinsulin biosynthesis in rat pancreatic islets. J Biol Chem. 1993;268(6):4276–4280. - PubMed
-
- Allen JR, Nguyen LX, Sargent KEG, Lipson KL, Hackett A, Urano F. High ER stress in β-cells stimulates intracellular degradation of misfolded insulin. Biochem Biophys Res Commun. 2004;324(1):166–170. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
