

Salinomycin and other polyether ionophores are a new class of antiscarring agent
- PMID: 25538236
- PMCID: PMC4319023
- DOI: 10.1074/jbc.M114.601872
Salinomycin and other polyether ionophores are a new class of antiscarring agent
Abstract
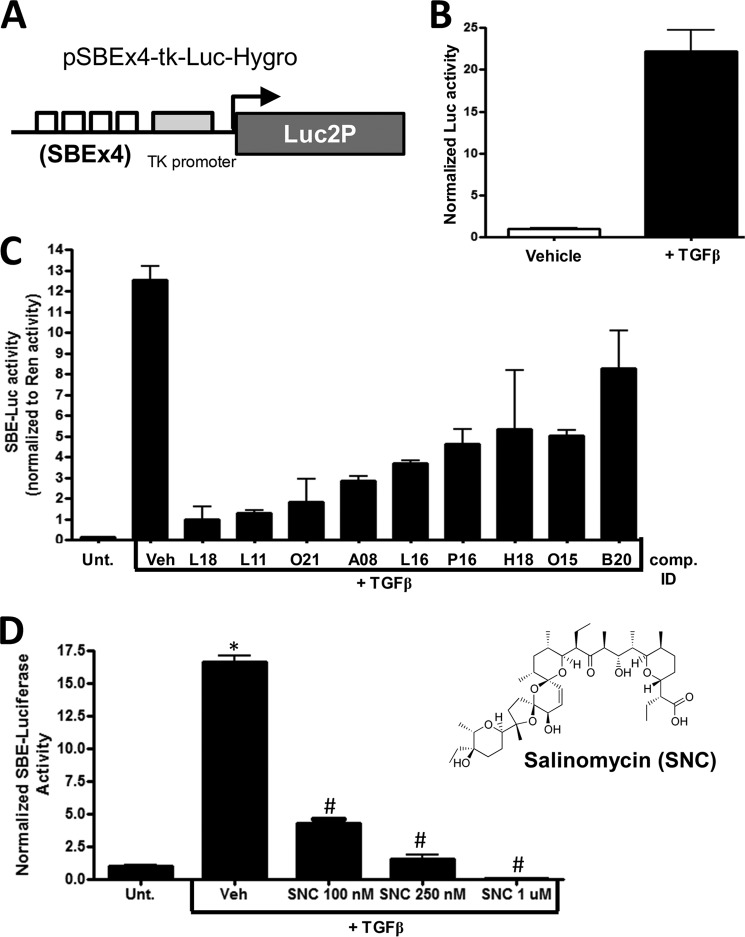
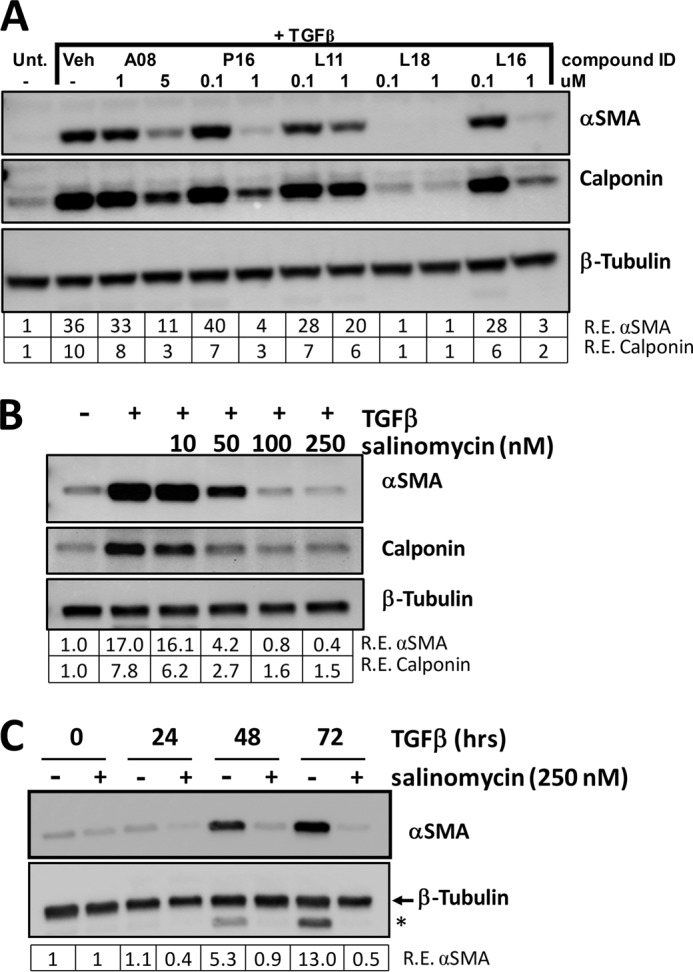
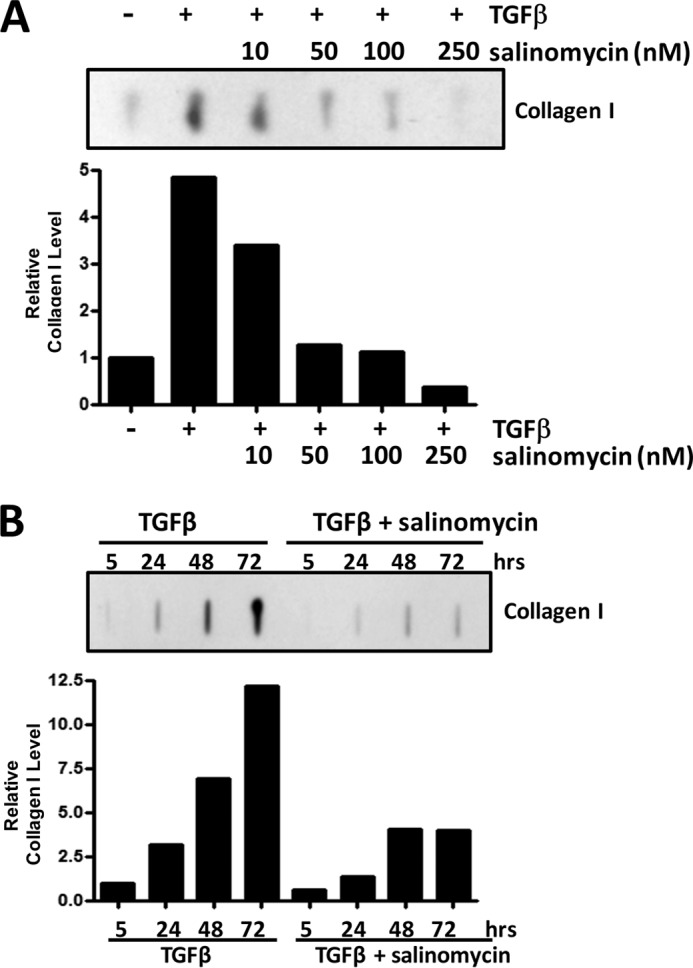
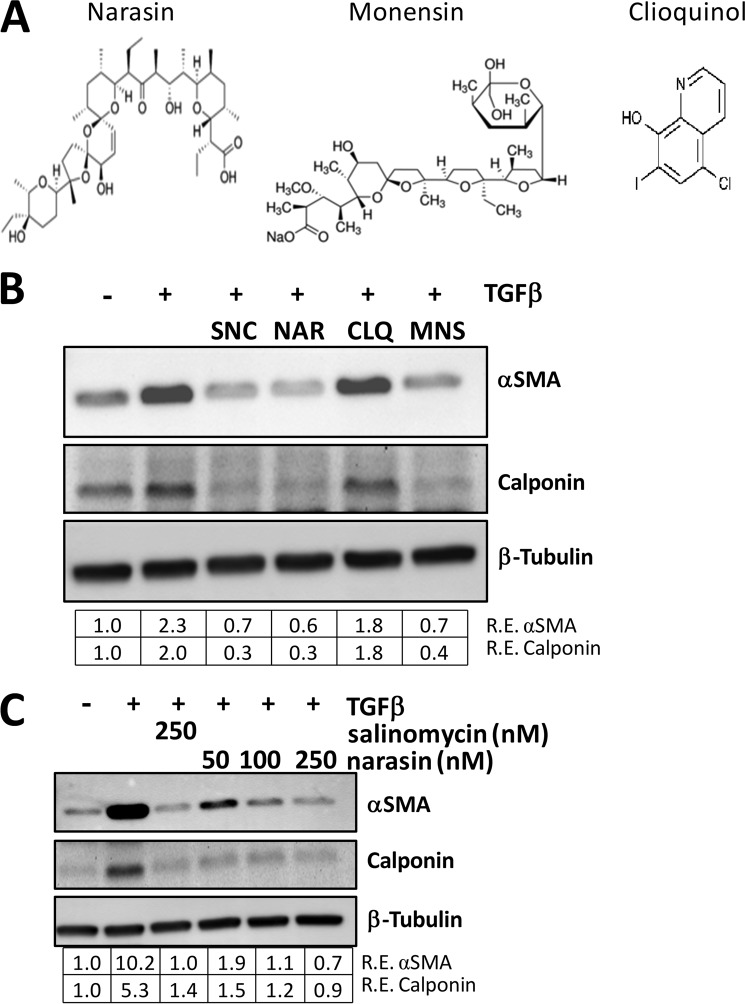
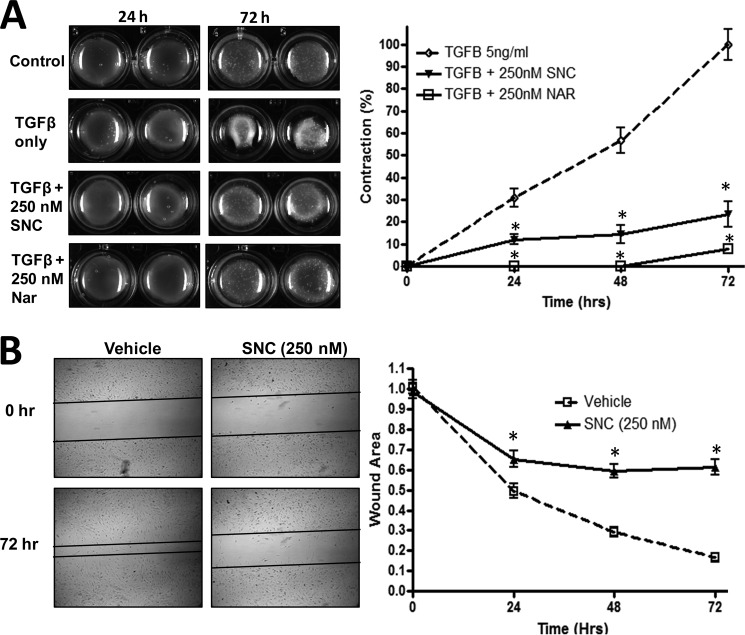
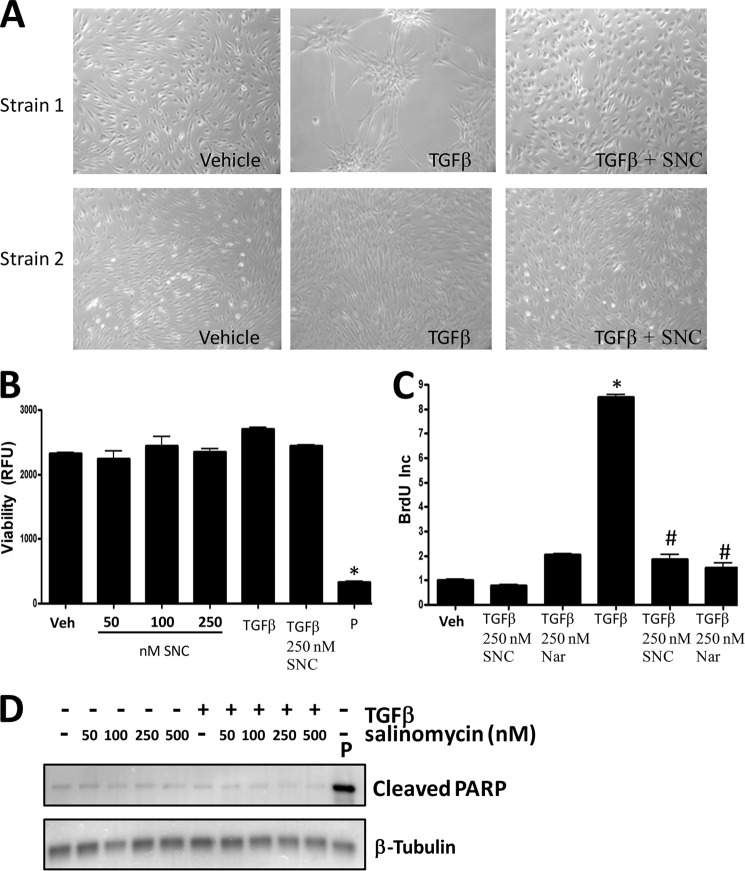
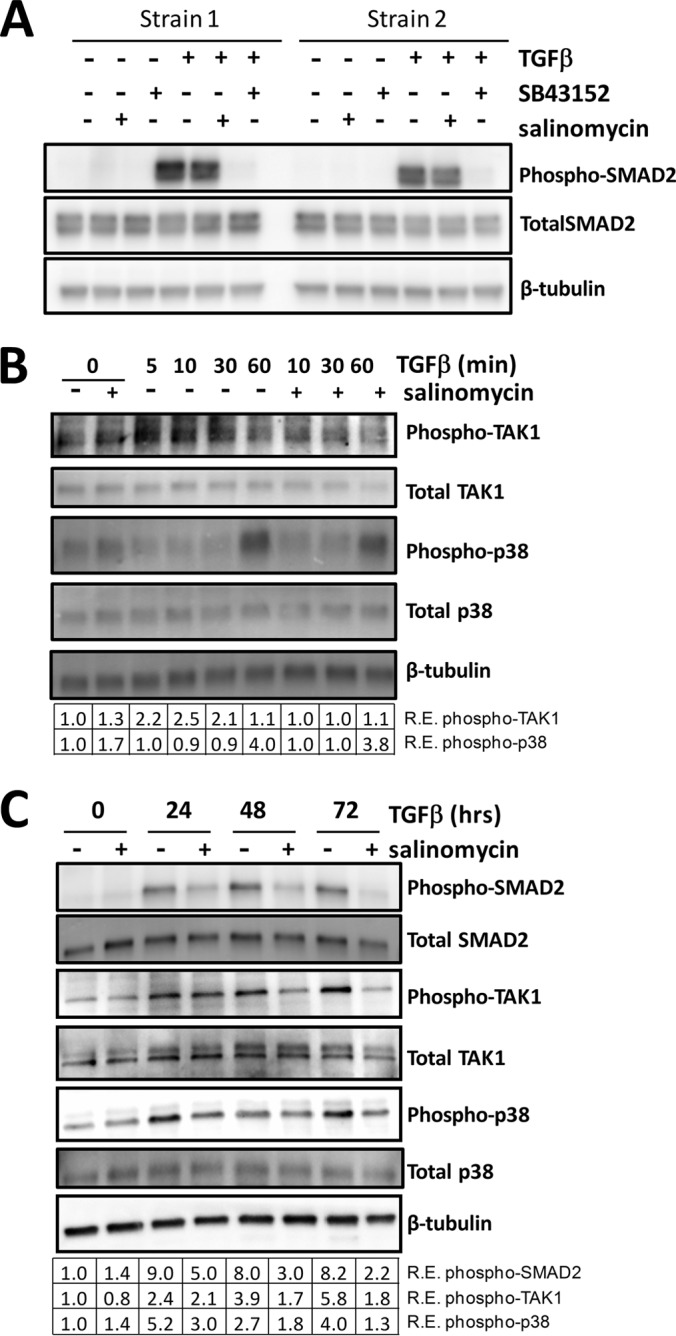
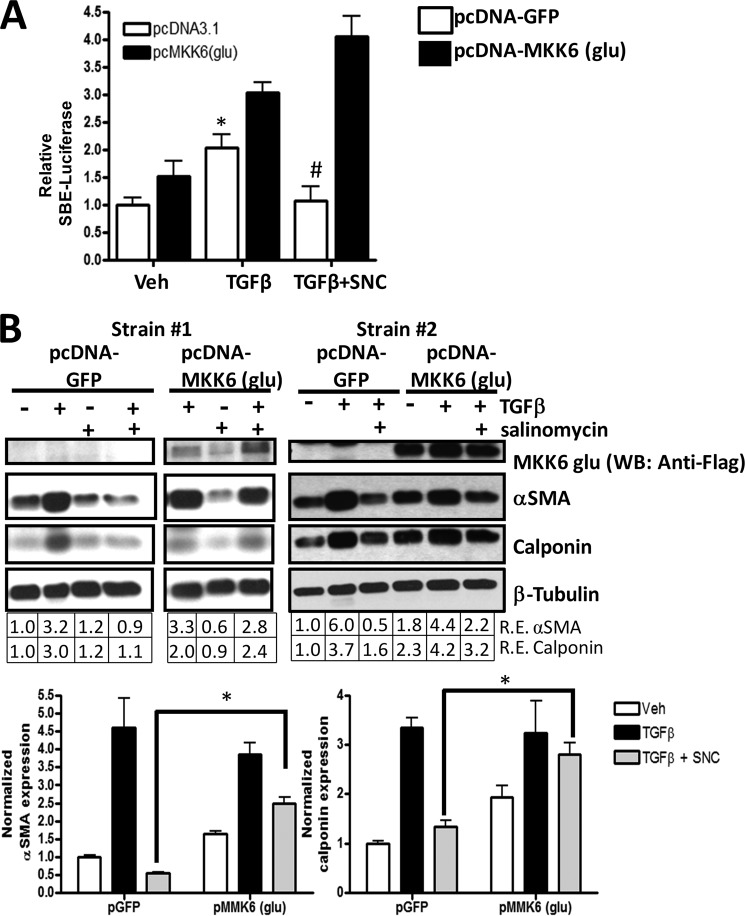
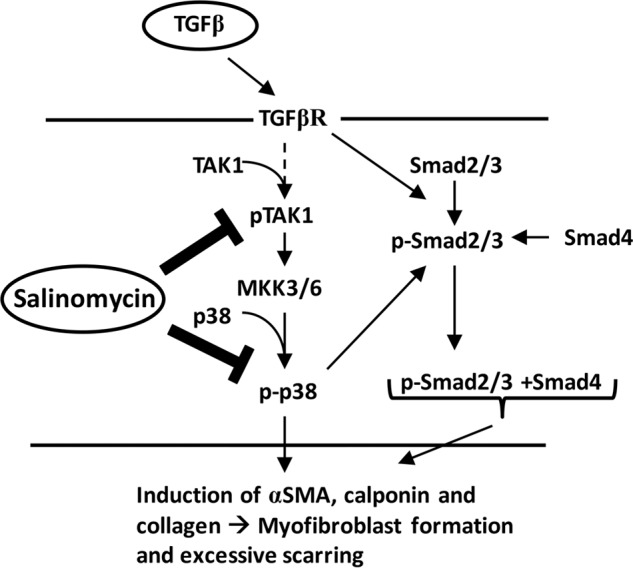
Although scarring is a component of wound healing, excessive scar formation is a debilitating condition that results in pain, loss of tissue function, and even death. Many tissues, including the lungs, heart, skin, and eyes, can develop excessive scar tissue as a result of tissue injury, chronic inflammation, or autoimmune disease. Unfortunately, there are few, if any, effective treatments to prevent excess scarring, and new treatment strategies are needed. Using HEK293FT cells stably transfected with a TGFβ-dependent luciferase reporter, we performed a small molecule screen to identify novel compounds with antiscarring activity. We discovered that the polyether ionophore salinomycin potently inhibited the formation of scar-forming myofibroblasts. Salinomycin (250 nm) blocked TGFβ-dependent expression of the cardinal myofibroblast products α smooth muscle actin, calponin, and collagen in primary human fibroblasts without causing cell death. Salinomycin blocked phosphorylation and activation of TAK1 and p38, two proteins fundamentally involved in signaling myofibroblast and scar formation. Expression of constitutively active mitogen activated kinase kinase 6, which activates p38 MAPK, attenuated the ability of salinomycin to block myofibroblast formation, demonstrating that salinomycin targets the p38 kinase pathway to disrupt TGFβ signaling. These data identify salinomycin and other polyether ionophores as novel potential antiscarring therapeutics.
Keywords: Collagen; Fibrosis; MKK6; Myofibroblast; Polyether Ionophores; SMAD Transcription Factor; Salinomycin; TGFβ; p38; p38 MAPK.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
FGF2-mediated attenuation of myofibroblast activation is modulated by distinct MAPK signaling pathways in human dermal fibroblasts.J Dermatol Sci. 2017 Dec;88(3):339-348. doi: 10.1016/j.jdermsci.2017.08.013. Epub 2017 Sep 1. J Dermatol Sci. 2017. PMID: 28899582 Free PMC article.
-
TRPV1 potentiates TGFβ-induction of corneal myofibroblast development through an oxidative stress-mediated p38-SMAD2 signaling loop.PLoS One. 2013 Oct 2;8(10):e77300. doi: 10.1371/journal.pone.0077300. eCollection 2013. PLoS One. 2013. PMID: 24098582 Free PMC article.
-
Transforming growth factor-beta receptor type 1 (TGFbetaRI) kinase activity but not p38 activation is required for TGFbetaRI-induced myofibroblast differentiation and profibrotic gene expression.Mol Pharmacol. 2006 Aug;70(2):518-31. doi: 10.1124/mol.105.021600. Epub 2006 May 17. Mol Pharmacol. 2006. PMID: 16707625
-
Modulation of Smad signaling by non-TGFβ components in myofibroblast generation during wound healing in corneal stroma.Exp Eye Res. 2016 Jan;142:40-8. doi: 10.1016/j.exer.2014.12.015. Exp Eye Res. 2016. PMID: 26675402 Review.
-
BH3 mimetics as anti-fibrotic therapy: Unleashing the mitochondrial pathway of apoptosis in myofibroblasts.Matrix Biol. 2018 Aug;68-69:94-105. doi: 10.1016/j.matbio.2018.01.020. Epub 2018 Jan 31. Matrix Biol. 2018. PMID: 29408011 Review.
Cited by
-
Evolution of a Biosynthetic Temporary Skin Substitute: A Preliminary Study.Eplasty. 2015 Jul 20;15:e30. eCollection 2015. Eplasty. 2015. PMID: 26229573 Free PMC article.
-
Protective transcriptional mechanisms in cardiomyocytes and cardiac fibroblasts.J Mol Cell Cardiol. 2019 Jul;132:1-12. doi: 10.1016/j.yjmcc.2019.04.023. Epub 2019 Apr 28. J Mol Cell Cardiol. 2019. PMID: 31042488 Free PMC article. Review.
-
Inhibition of p38 mitogen-activated protein kinases may attenuate scar proliferation after cleft lip surgery in rabbits via Smads signaling pathway.Eur J Med Res. 2022 Jul 20;27(1):126. doi: 10.1186/s40001-022-00757-1. Eur J Med Res. 2022. PMID: 35858881 Free PMC article.
-
In Vitro Characterization of Variable Porosity Wound Dressing With Anti-Scar Properties.Eplasty. 2018 May 25;18:e21. eCollection 2018. Eplasty. 2018. PMID: 29896321 Free PMC article.
-
Ionizing radiation induces myofibroblast differentiation via lactate dehydrogenase.Am J Physiol Lung Cell Mol Physiol. 2015 Oct 15;309(8):L879-87. doi: 10.1152/ajplung.00153.2015. Epub 2015 Aug 7. Am J Physiol Lung Cell Mol Physiol. 2015. PMID: 26254422 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
