

Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies
- PMID: 25524009
- PMCID: PMC4411703
- DOI: 10.1186/s12881-014-0139-9
Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies
Abstract
Background: DAVID syndrome is a rare condition combining anterior pituitary hormone deficiency with common variable immunodeficiency. NFKB2 mutations have recently been identified in patients with ACTH and variable immunodeficiency. A similar mutation was previously found in Nfkb2 in the immunodeficient Lym1 mouse strain, but the effect of the mutation on endocrine function was not evaluated.
Methods: We ascertained six unrelated DAVID syndrome families. We performed whole exome and traditional Sanger sequencing to search for causal genes. Lym1 mice were examined for endocrine developmental anomalies.
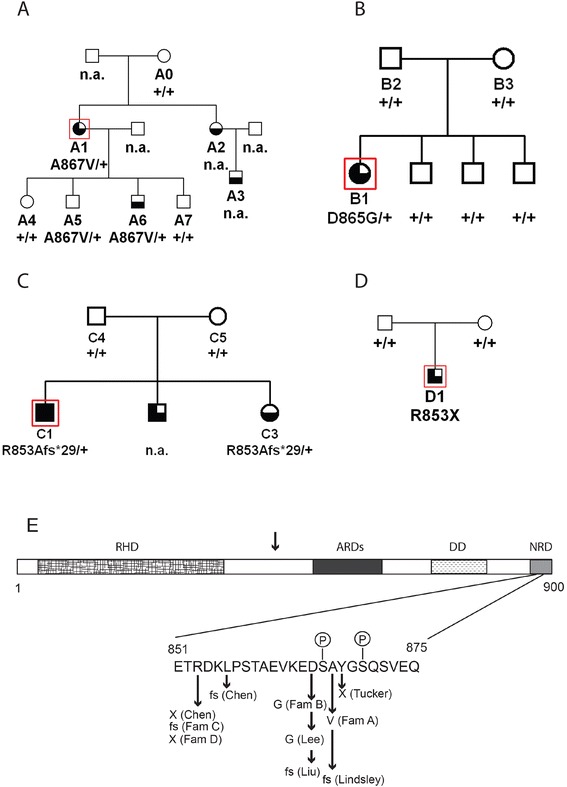
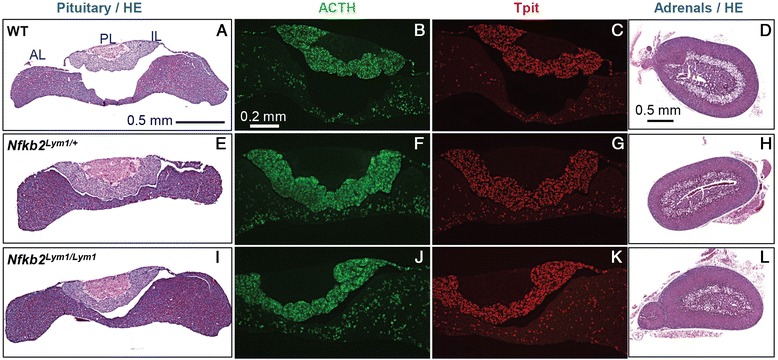
Results: Mutations in the NFKB2 gene were identified in three of our families through whole exome sequencing, and in a fourth by direct Sanger sequencing. De novo origin of the mutations could be demonstrated in three of the families. All mutations lie near the C-terminus of the protein-coding region, near signals required for processing of NFΚB2 protein by the alternative pathway. Two of the probands had anatomical pituitary anomalies, and one had growth and thyroid hormone as well as ACTH deficiency; these findings have not been previously reported. Two children of one of the probands carried the mutation and have to date exhibited only an immune phenotype. No mutations were found near the C-terminus of NFKB2 in the remaining two probands; whole exome sequencing has been performed for one of these. Lym1 mice, carrying a similar Nfkb2 C-terminal mutation, showed normal pituitary anatomy and expression of proopiomelanocortin (POMC).
Conclusions: We confirm previous findings that mutations near the C-terminus of NFKB2 cause combined endocrine and immunodeficiencies. De novo status of the mutations was confirmed in all cases for which both parents were available. The mutations are consistent with a dominant gain-of-function effect, generating an unprocessed NFKB2 super-repressor protein. We expand the potential phenotype of such NFKB2 mutations to include additional pituitary hormone deficiencies as well as anatomical pituitary anomalies. The lack of an observable endocrine phenotype in Lym1 mice suggests that the endocrine component of DAVID syndrome is either not due to a direct role of NFKB pathways on pituitary development, or else that human and mouse pituitary development differ in its requirements for NFKB pathway function.
Figures
Similar articles
-
Clinical and Immunological Phenotype of Patients With Primary Immunodeficiency Due to Damaging Mutations in NFKB2.Front Immunol. 2019 Mar 19;10:297. doi: 10.3389/fimmu.2019.00297. eCollection 2019. Front Immunol. 2019. PMID: 30941118 Free PMC article.
-
Deficient anterior pituitary with common variable immune deficiency (DAVID syndrome): a new case and literature reports.J Neuroendocrinol. 2023 Jun;35(6):e13287. doi: 10.1111/jne.13287. Epub 2023 Jun 15. J Neuroendocrinol. 2023. PMID: 37322808 Review.
-
Central Diabetes Insipidus in a Patient With NFKB2 Mutation: Expanding the Endocrine Phenotype in DAVID Syndrome.J Clin Endocrinol Metab. 2019 Sep 1;104(9):4051-4057. doi: 10.1210/jc.2019-00469. J Clin Endocrinol Metab. 2019. PMID: 31150062
-
Combined immune deficiency in a patient with a novel NFKB2 mutation.J Clin Immunol. 2014 Nov;34(8):910-5. doi: 10.1007/s10875-014-0095-3. Epub 2014 Sep 10. J Clin Immunol. 2014. PMID: 25205549 Free PMC article.
-
NFKB2 mutation in common variable immunodeficiency and isolated adrenocorticotropic hormone deficiency: A case report and review of literature.Medicine (Baltimore). 2016 Oct;95(40):e5081. doi: 10.1097/MD.0000000000005081. Medicine (Baltimore). 2016. PMID: 27749582 Free PMC article. Review.
Cited by
-
Inherited human RelB deficiency impairs innate and adaptive immunity to infection.Proc Natl Acad Sci U S A. 2024 Sep 10;121(37):e2321794121. doi: 10.1073/pnas.2321794121. Epub 2024 Sep 4. Proc Natl Acad Sci U S A. 2024. PMID: 39231201 Free PMC article.
-
Beyond monogenetic rare variants: tackling the low rate of genetic diagnoses in predominantly antibody deficiency.Cell Mol Immunol. 2021 Mar;18(3):588-603. doi: 10.1038/s41423-020-00520-8. Epub 2020 Aug 17. Cell Mol Immunol. 2021. PMID: 32801365 Free PMC article. Review.
-
Case Report: A child with NFKB1 haploinsufficiency explaining the linkage between immunodeficiency and short stature.Front Immunol. 2023 Aug 3;14:1224603. doi: 10.3389/fimmu.2023.1224603. eCollection 2023. Front Immunol. 2023. PMID: 37600787 Free PMC article.
-
Novel nonsense gain-of-function NFKB2 mutations associated with a combined immunodeficiency phenotype.Blood. 2017 Sep 28;130(13):1553-1564. doi: 10.1182/blood-2017-05-782177. Epub 2017 Aug 4. Blood. 2017. PMID: 28778864 Free PMC article.
-
Late-Onset Isolated Corticotrope Deficiency in a Woman with Down Syndrome.Case Rep Endocrinol. 2021 Apr 27;2021:5562831. doi: 10.1155/2021/5562831. eCollection 2021. Case Rep Endocrinol. 2021. PMID: 34007493 Free PMC article.
References
-
- Quentien MH, Delemer B, Papadimitriou DT, Souchon PF, Jaussaud R, Pagnier A, Munzer M, Jullien N, Reynaud R, Galon-Faure N, Enjalbert A, Barlier A, Brue T. Deficit in anterior pituitary function and variable immune deficiency (DAVID) in children presenting with adrenocorticotropin deficiency and severe infections. J Clin Endocrinol Metab. 2012;97(1):E121–E128. doi: 10.1210/jc.2011-0407. - DOI - PubMed
-
- Chen K, Coonrod EM, Kumanovics A, Franks ZF, Durtschi JD, Margraf RL, Wu W, Heikal NM, Augustine NH, Ridge PG, Hill HR, Jorde LB, Weyrich AS, Zimmerman GA, Gundlapalli AV, Bohnsack JF, Voelkerding KV. Germline mutations in NFKB2 implicate the noncanonical NF-kappaB pathway in the pathogenesis of common variable immunodeficiency. Am J Hum Genet. 2013;93(5):812–824. doi: 10.1016/j.ajhg.2013.09.009. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
