

HSP47 and FKBP65 cooperate in the synthesis of type I procollagen
- PMID: 25510505
- PMCID: PMC4355024
- DOI: 10.1093/hmg/ddu608
HSP47 and FKBP65 cooperate in the synthesis of type I procollagen
Abstract
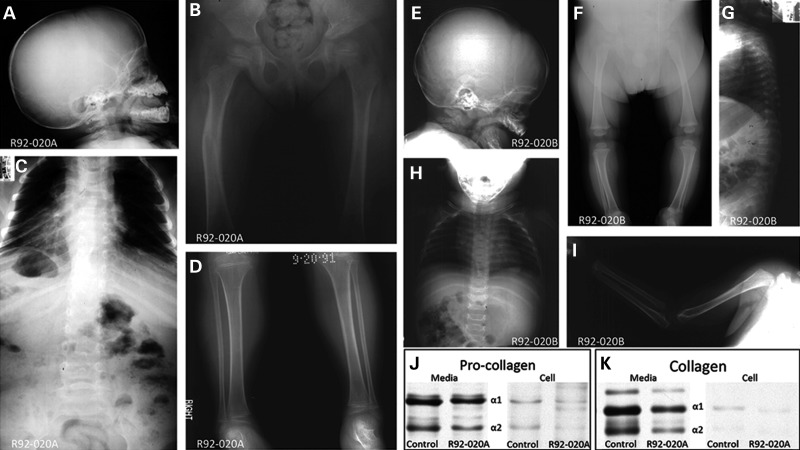
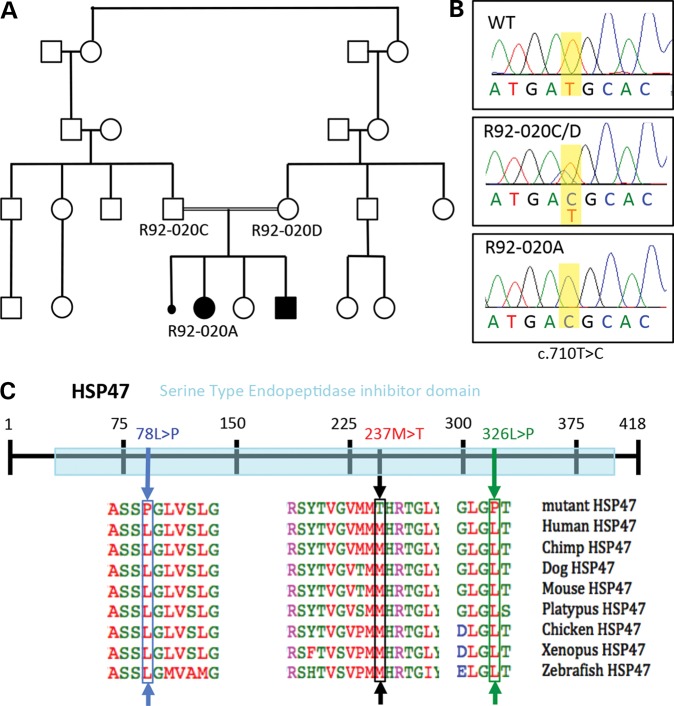
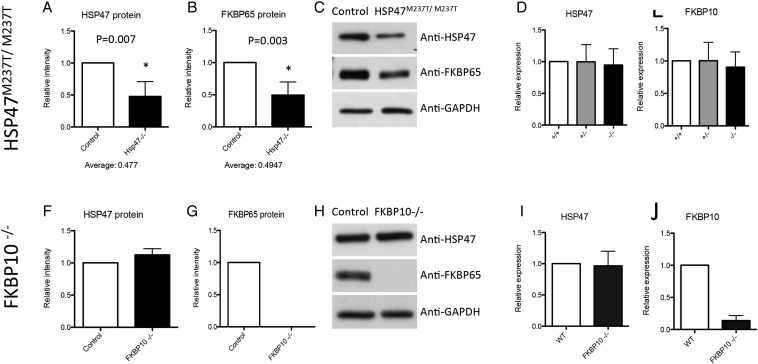
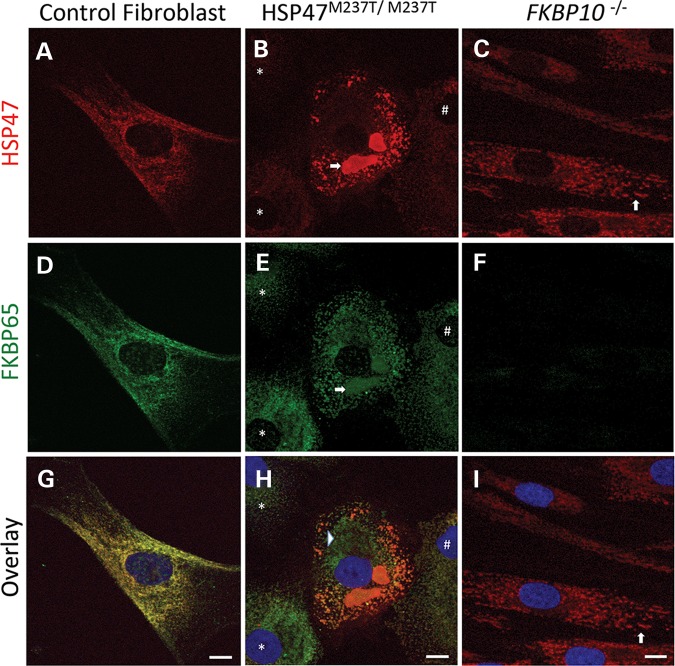
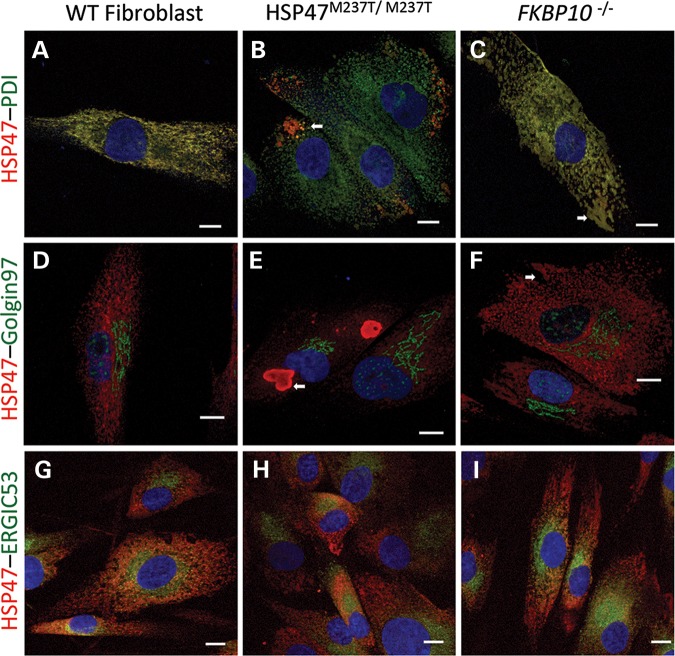
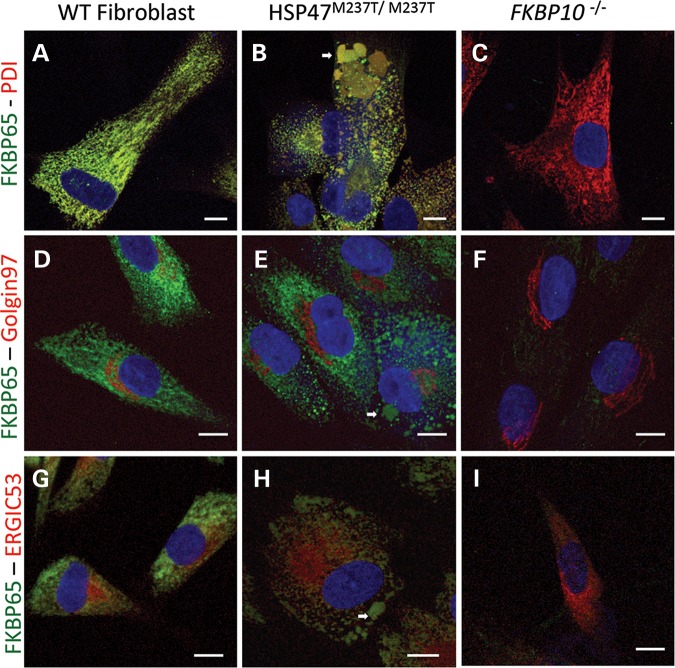
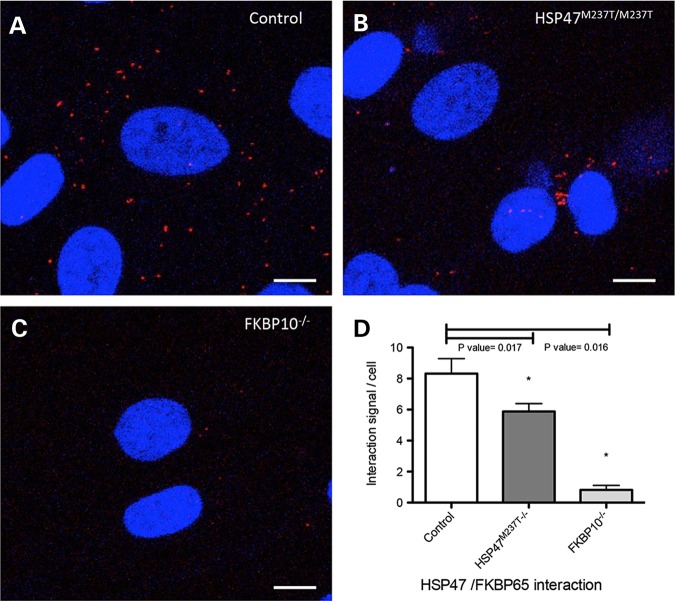
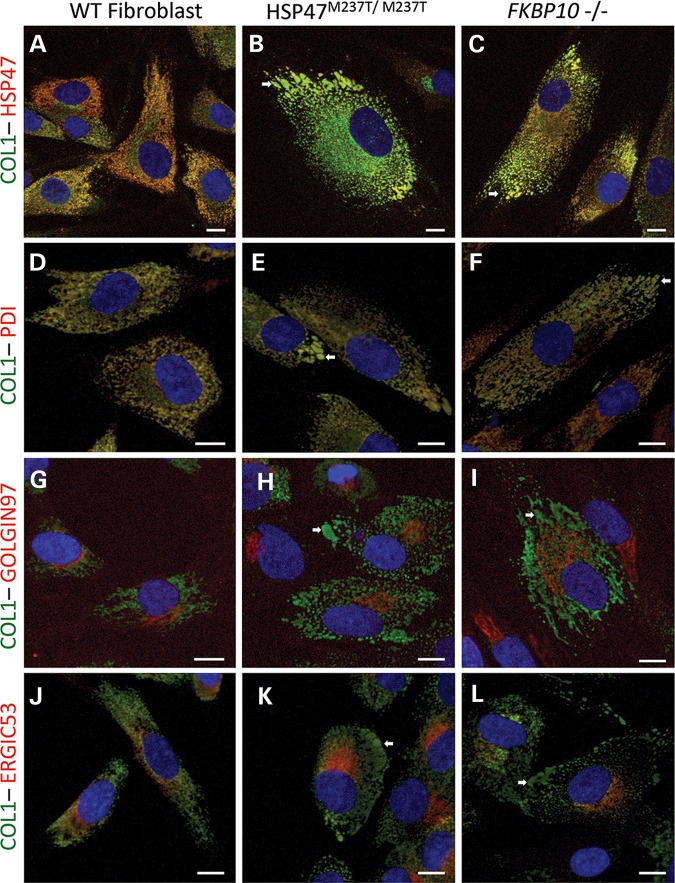
Osteogenesis imperfecta (OI) is a genetic disorder that results in low bone mineral density and brittle bones. Most cases result from dominant mutations in the type I procollagen genes, but mutations in a growing number of genes have been identified that produce autosomal recessive forms of the disease. Among these include mutations in the genes SERPINH1 and FKBP10, which encode the type I procollagen chaperones HSP47 and FKBP65, respectively, and predominantly produce a moderately severe form of OI. Little is known about the biochemical consequences of the mutations and how they produce OI. We have identified a new OI mutation in SERPINH1 that results in destabilization and mislocalization of HSP47 and secondarily has similar effects on FKBP65. We found evidence that HSP47 and FKBP65 act cooperatively during posttranslational maturation of type I procollagen and that FKBP65 and HSP47 but fail to properly interact in mutant HSP47 cells. These results thus reveal a common cellular pathway in cases of OI caused by HSP47 and FKBP65 deficiency.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Heat shock protein 47 and 65-kDa FK506-binding protein weakly but synergistically interact during collagen folding in the endoplasmic reticulum.J Biol Chem. 2017 Oct 20;292(42):17216-17224. doi: 10.1074/jbc.M117.802298. Epub 2017 Aug 31. J Biol Chem. 2017. PMID: 28860186 Free PMC article.
-
Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta.Am J Hum Genet. 2010 Mar 12;86(3):389-98. doi: 10.1016/j.ajhg.2010.01.034. Epub 2010 Feb 25. Am J Hum Genet. 2010. PMID: 20188343 Free PMC article.
-
Interaction between KDELR2 and HSP47 as a Key Determinant in Osteogenesis Imperfecta Caused by Bi-allelic Variants in KDELR2.Am J Hum Genet. 2020 Nov 5;107(5):989-999. doi: 10.1016/j.ajhg.2020.09.009. Epub 2020 Oct 13. Am J Hum Genet. 2020. PMID: 33053334 Free PMC article.
-
Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta.Calcif Tissue Int. 2013 Oct;93(4):338-47. doi: 10.1007/s00223-013-9723-9. Epub 2013 Mar 19. Calcif Tissue Int. 2013. PMID: 23508630 Free PMC article. Review.
-
Recessively inherited forms of osteogenesis imperfecta.Annu Rev Genet. 2012;46:475-97. doi: 10.1146/annurev-genet-110711-155608. Annu Rev Genet. 2012. PMID: 23145505 Review.
Cited by
-
Genetic causes and mechanisms of Osteogenesis Imperfecta.Bone. 2017 Sep;102:40-49. doi: 10.1016/j.bone.2017.02.004. Epub 2017 Feb 15. Bone. 2017. PMID: 28232077 Free PMC article. Review.
-
COL3A1, COL6A3, and SERPINH1 Are Related to Glucocorticoid-Induced Osteoporosis Occurrence According to Integrated Bioinformatics Analysis.Med Sci Monit. 2020 Oct 1;26:e925474. doi: 10.12659/MSM.925474. Med Sci Monit. 2020. PMID: 32999266 Free PMC article.
-
SERPINH1 is a Potential Prognostic Biomarker and Correlated With Immune Infiltration: A Pan-Cancer Analysis.Front Genet. 2022 Jan 4;12:756094. doi: 10.3389/fgene.2021.756094. eCollection 2021. Front Genet. 2022. PMID: 35058967 Free PMC article.
-
R-catcher, a potent molecular tool to unveil the arginylome.Cell Mol Life Sci. 2021 Apr;78(7):3725-3741. doi: 10.1007/s00018-021-03805-x. Epub 2021 Mar 9. Cell Mol Life Sci. 2021. PMID: 33687501 Free PMC article.
-
Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease.J Biol Chem. 2019 Feb 8;294(6):2133-2141. doi: 10.1074/jbc.TM118.002812. Epub 2018 Dec 12. J Biol Chem. 2019. PMID: 30541925 Free PMC article. Review.
References
-
- Murakami T., Saito A., Hino S.I., Kondo S., Kanemoto S., Chihara K., Sekiya H., Tsumagari K., Ochiai K., Yoshinaga K., et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 2009;10:1205–1211. - PubMed
-
- Byers P.H., Pyott S.M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 2012;46:475–497. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
