

CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation
- PMID: 25476776
- PMCID: PMC4423185
- DOI: 10.1038/cdd.2014.194
CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation
Abstract
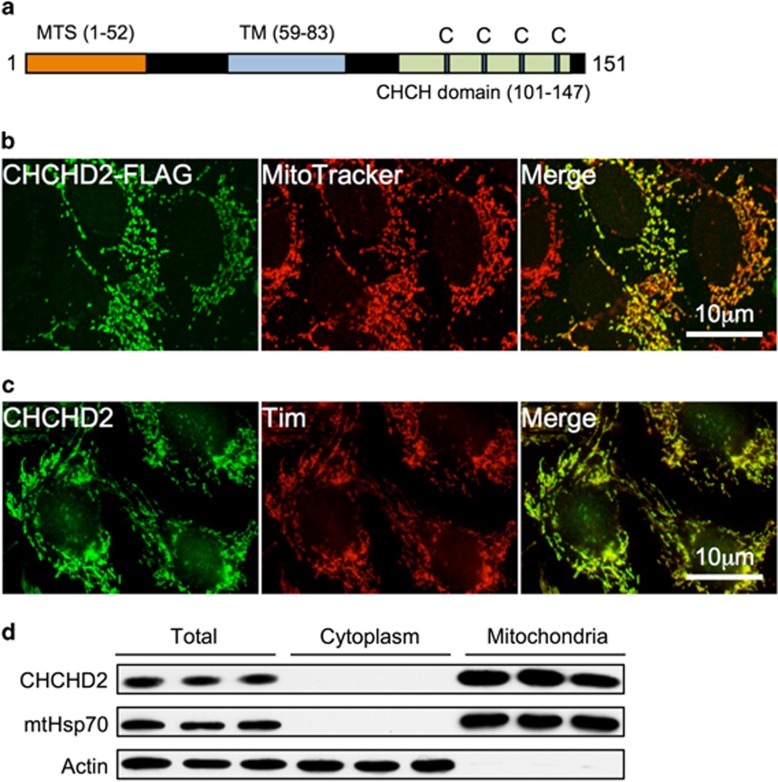
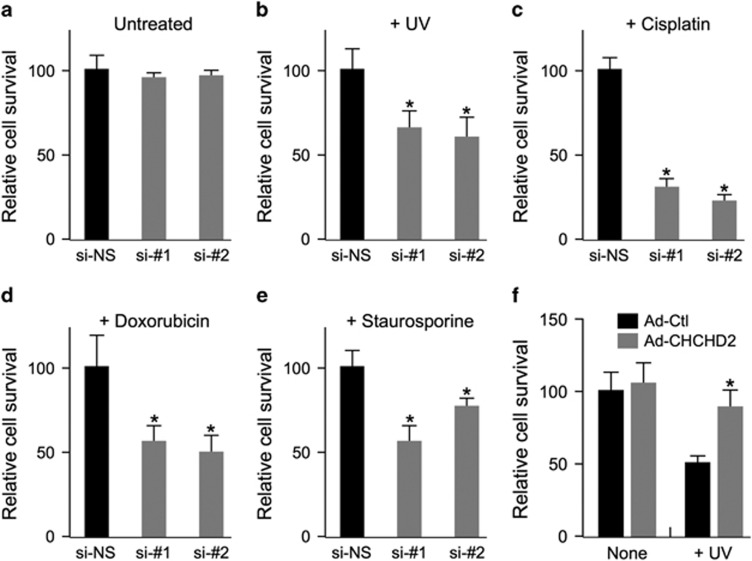
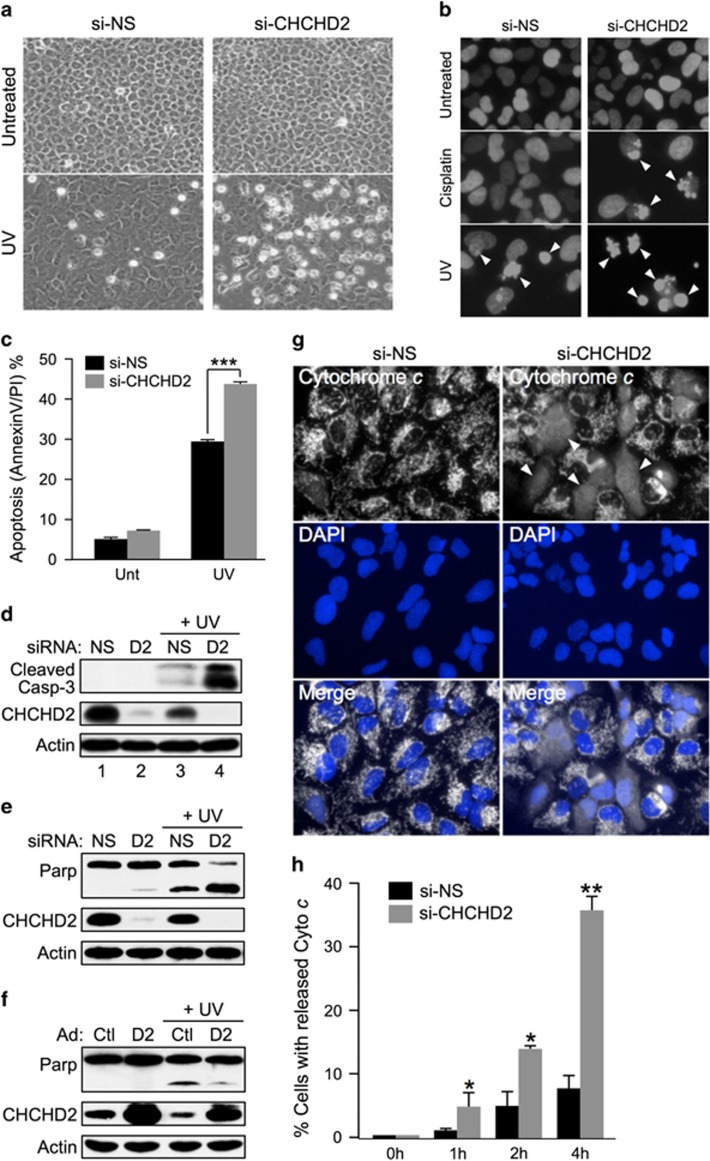
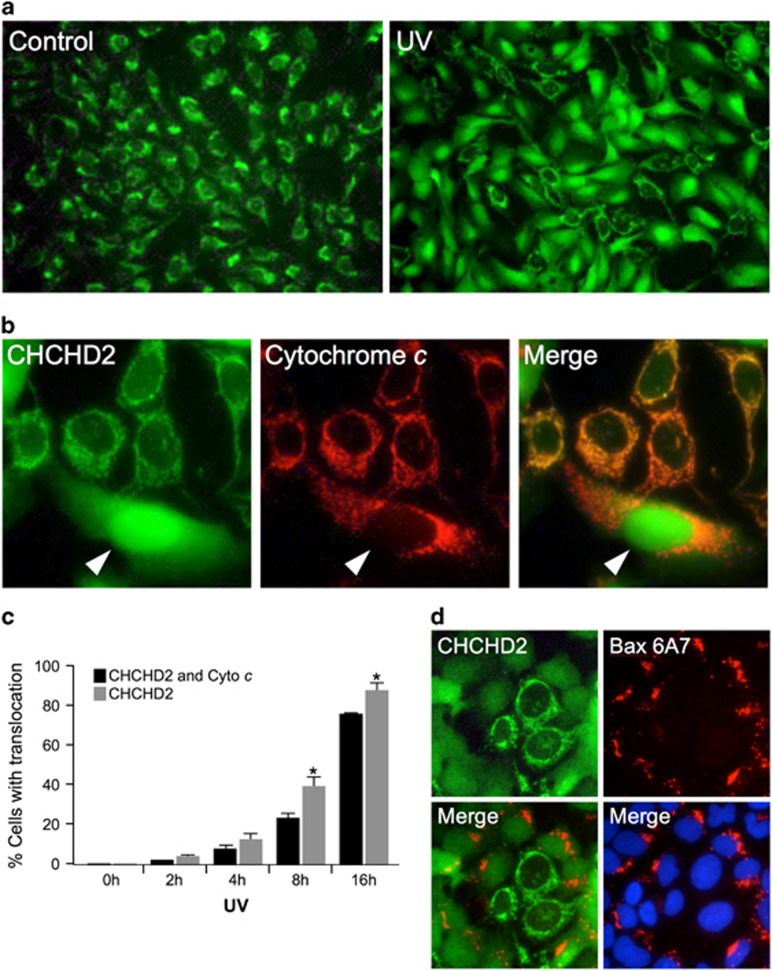
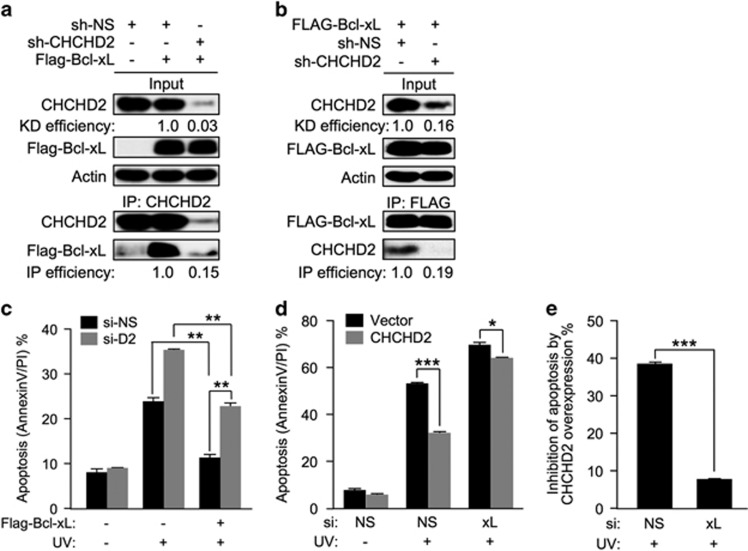
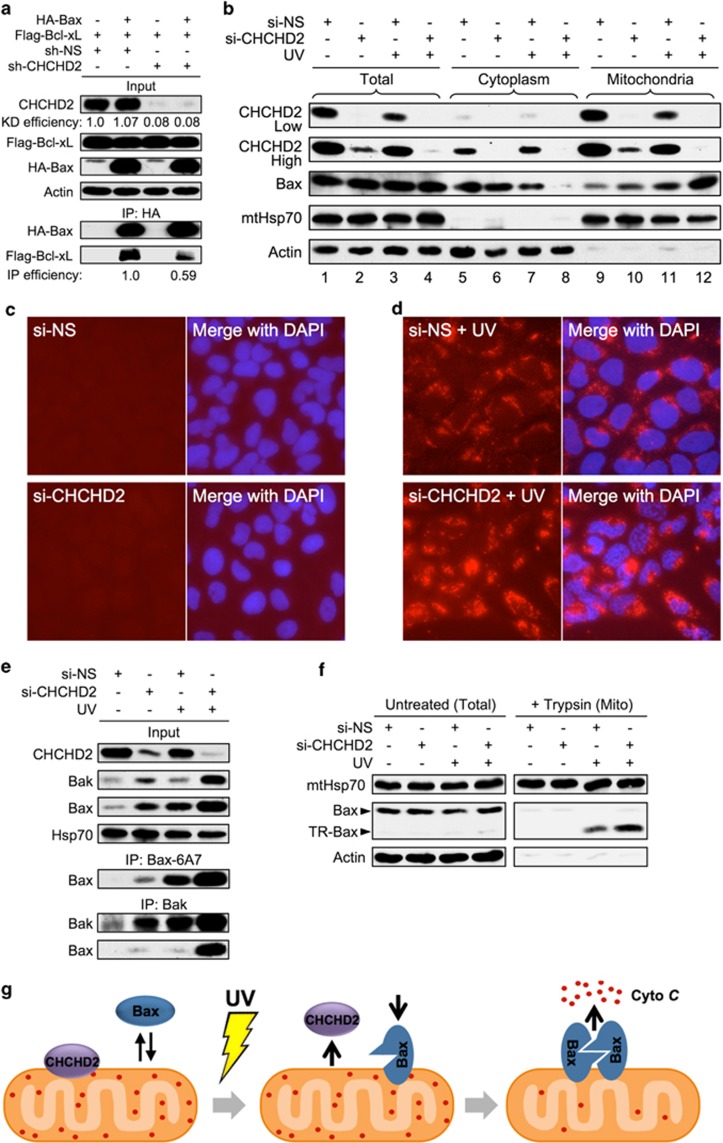
Mitochondrial outer membrane permeabilization (MOMP) is a critical control point during apoptosis that results in the release of pro-apoptotic mitochondrial contents such as cytochrome c. MOMP is largely controlled by Bcl-2 family proteins such as Bax, which under various apoptotic stresses becomes activated and oligomerizes on the outer mitochondrial membrane. Bax oligomerization helps promote the diffusion of the mitochondrial contents into the cytoplasm activating the caspase cascade. In turn, Bax is regulated primarily by anti-apoptotic Bcl-2 proteins including Bcl-xL, which was recently shown to prevent Bax from accumulating at the mitochondria. However, the exact mechanisms by which Bcl-xL regulates Bax and thereby MOMP remain partially understood. In this study, we show that the small CHCH-domain-containing protein CHCHD2 binds to Bcl-xL and inhibits the mitochondrial accumulation and oligomerization of Bax. Our data show that in response to apoptotic stimuli, mitochondrial CHCHD2 decreases prior to MOMP. Furthermore, when CHCHD2 is absent from the mitochondria, the ability of Bcl-xL to inhibit Bax activation and to prevent apoptosis is attenuated, which results in increases in Bax oligomerization, MOMP and apoptosis. Collectively, our findings establish CHCHD2, a previously uncharacterized small mitochondrial protein with no known homology to the Bcl-2 family, as one of the negative regulators of mitochondria-mediated apoptosis.
Figures
Similar articles
-
The N-terminus and alpha-5, alpha-6 helices of the pro-apoptotic protein Bax, modulate functional interactions with the anti-apoptotic protein Bcl-xL.BMC Cell Biol. 2007 May 23;8:16. doi: 10.1186/1471-2121-8-16. BMC Cell Biol. 2007. PMID: 17519046 Free PMC article.
-
Bax/Bak-dependent, Drp1-independent Targeting of X-linked Inhibitor of Apoptosis Protein (XIAP) into Inner Mitochondrial Compartments Counteracts Smac/DIABLO-dependent Effector Caspase Activation.J Biol Chem. 2015 Sep 4;290(36):22005-18. doi: 10.1074/jbc.M115.643064. Epub 2015 Jul 1. J Biol Chem. 2015. PMID: 26134559 Free PMC article.
-
Bax is essential for Drp1-mediated mitochondrial fission but not for mitochondrial outer membrane permeabilization caused by photodynamic therapy.J Cell Physiol. 2011 Feb;226(2):530-41. doi: 10.1002/jcp.22362. J Cell Physiol. 2011. PMID: 20683914
-
Discoveries and controversies in BCL-2 protein-mediated apoptosis.FEBS J. 2016 Jul;283(14):2690-700. doi: 10.1111/febs.13527. Epub 2015 Oct 27. FEBS J. 2016. PMID: 26411300 Review.
-
Regulation of Bax mitochondrial localization by Bcl-2 and Bcl-x(L): keep your friends close but your enemies closer.Int J Biochem Cell Biol. 2013 Jan;45(1):64-7. doi: 10.1016/j.biocel.2012.09.022. Epub 2012 Oct 11. Int J Biochem Cell Biol. 2013. PMID: 23064052 Review.
Cited by
-
pS421 huntingtin modulates mitochondrial phenotypes and confers neuroprotection in an HD hiPSC model.Cell Death Dis. 2020 Sep 25;11(9):809. doi: 10.1038/s41419-020-02983-z. Cell Death Dis. 2020. PMID: 32978366 Free PMC article.
-
CHCHD2 up-regulation in Huntington disease mediates a compensatory protective response against oxidative stress.Cell Death Dis. 2024 Feb 10;15(2):126. doi: 10.1038/s41419-024-06523-x. Cell Death Dis. 2024. PMID: 38341417 Free PMC article.
-
Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders.Neurology. 2015 Dec 8;85(23):2016-25. doi: 10.1212/WNL.0000000000002170. Epub 2015 Nov 11. Neurology. 2015. PMID: 26561290 Free PMC article.
-
CHCHD2 Regulates Mitochondrial Function and Apoptosis of Ectopic Endometrial Stromal Cells in the Pathogenesis of Endometriosis.Reprod Sci. 2022 Aug;29(8):2152-2164. doi: 10.1007/s43032-021-00831-9. Epub 2022 Feb 14. Reprod Sci. 2022. PMID: 35157262
-
CHCHD2 is a potential prognostic factor for NSCLC and is associated with HIF-1a expression.BMC Pulm Med. 2020 Feb 13;20(1):40. doi: 10.1186/s12890-020-1079-0. BMC Pulm Med. 2020. PMID: 32054470 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
