

Synergistic antitumor interactions between MK-1775 and panobinostat in preclinical models of pancreatic cancer
- PMID: 25458954
- PMCID: PMC4282784
- DOI: 10.1016/j.canlet.2014.10.015
Synergistic antitumor interactions between MK-1775 and panobinostat in preclinical models of pancreatic cancer
Abstract
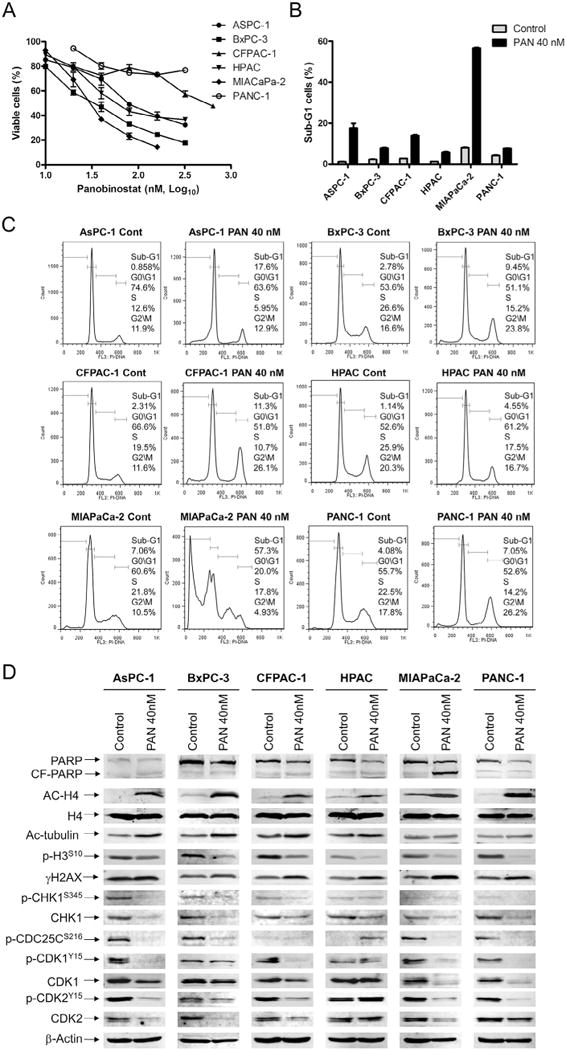
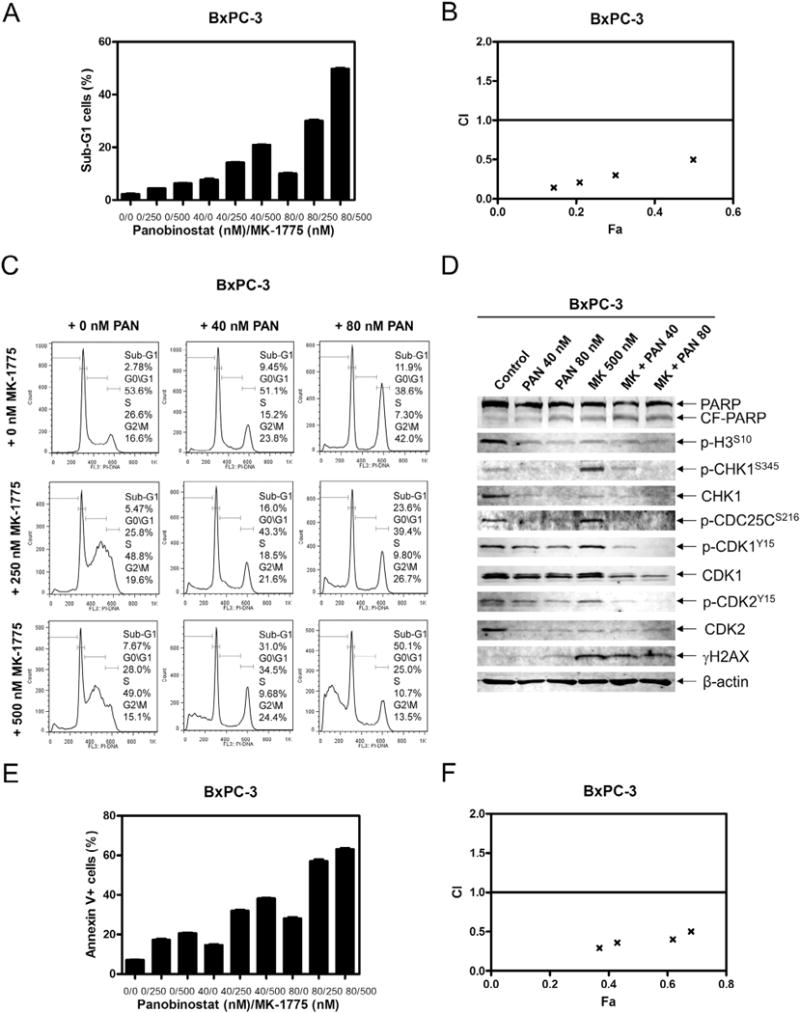
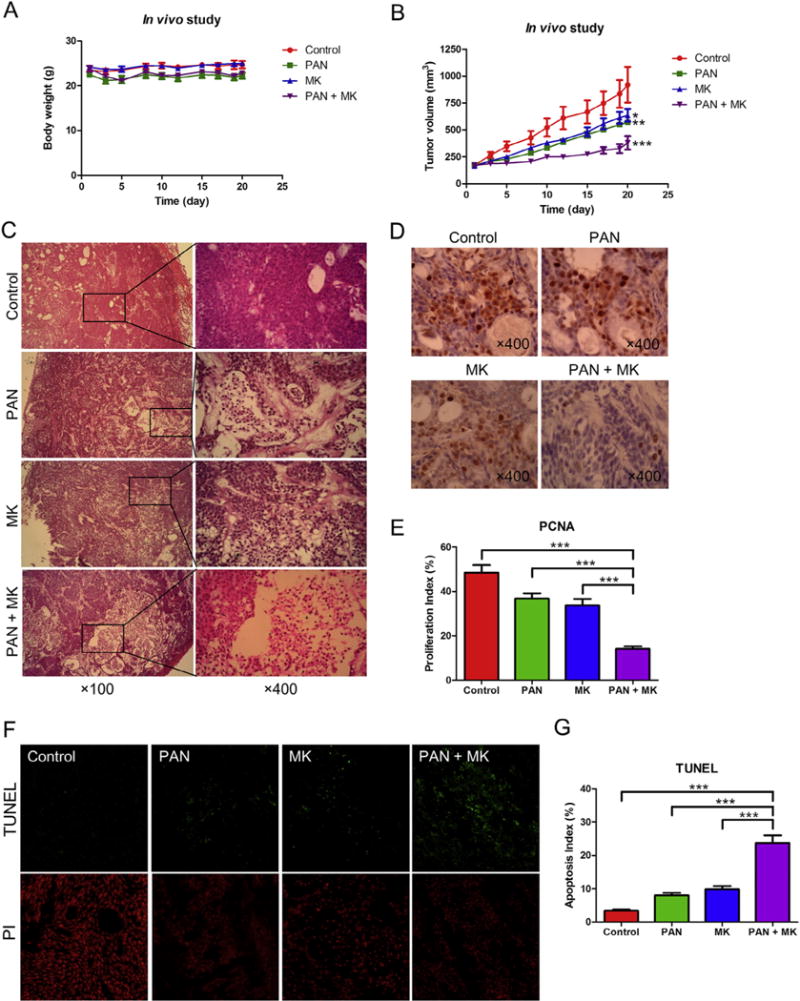
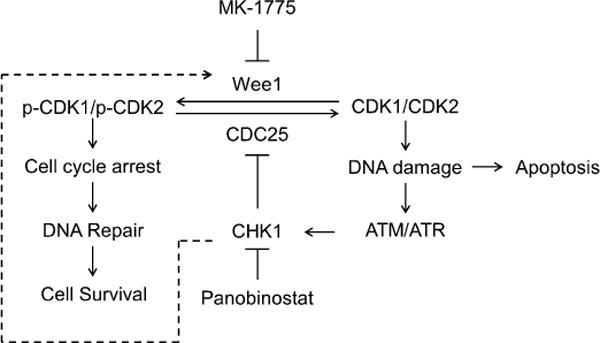
Pancreatic cancer remains a clinical challenge, thus new therapies are urgently needed. The selective Wee1 inhibitor MK-1775 has demonstrated promising results when combined with DNA damaging agents, and more recently with CHK1 inhibitors in various malignancies. We have previously demonstrated that treatment with the pan-histone deacetylase inhibitor panobinostat (LBH589) can cause down-regulation of CHK1. Accordingly, we investigated using panobinostat to down-regulate CHK1 in combination with MK-1775 to enhance cell death in preclinical pancreatic cancer models. We demonstrate that MK-1775 treatment results in increased H2AX phosphorylation, indicating increased DNA double-strand breaks, and activation of CHK1, which are both dependent on CDK activity. Combination of MK-1775 and panobinostat resulted in synergistic antitumor activity in six pancreatic cancer cell lines. Finally, our in vivo study using a pancreatic xenograft model reveals promising cooperative antitumor activity between MK-1775 and panobinostat. Our study provides compelling evidence that the combination of MK-1775 and panobinostat has antitumor activity in preclinical models of pancreatic cancer and supports the clinical development of panobinostat in combination with MK-1775 for the treatment of this deadly disease.
Keywords: CHK1; Drug combination; MK-1775; Pancreatic cancer; Panobinostat.
Copyright © 2014 Elsevier Ireland Ltd. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
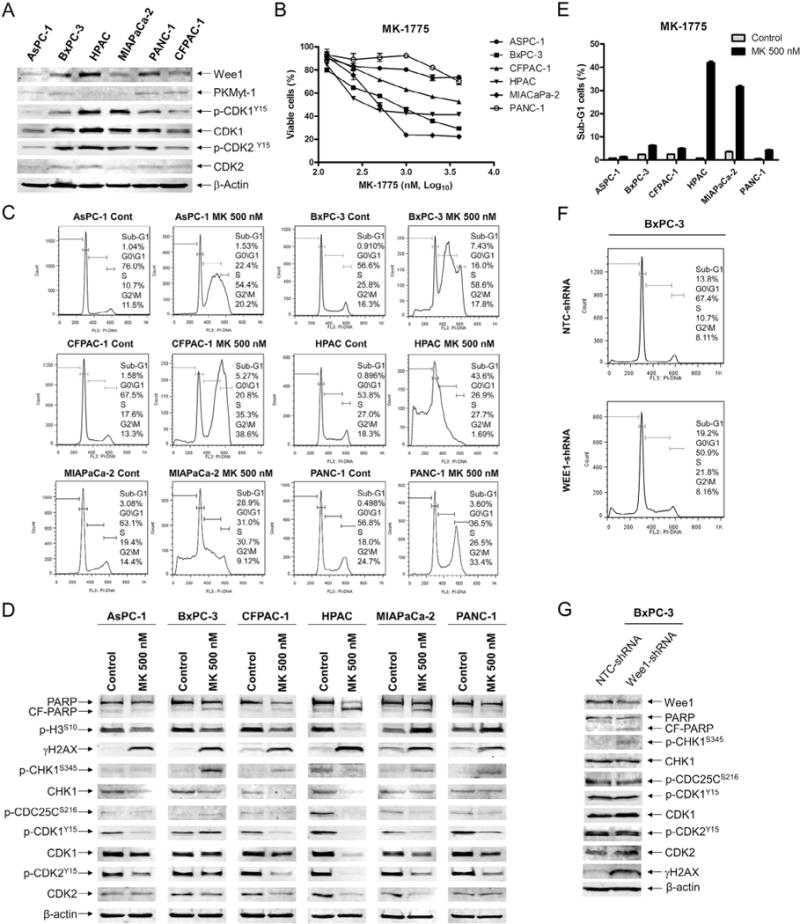
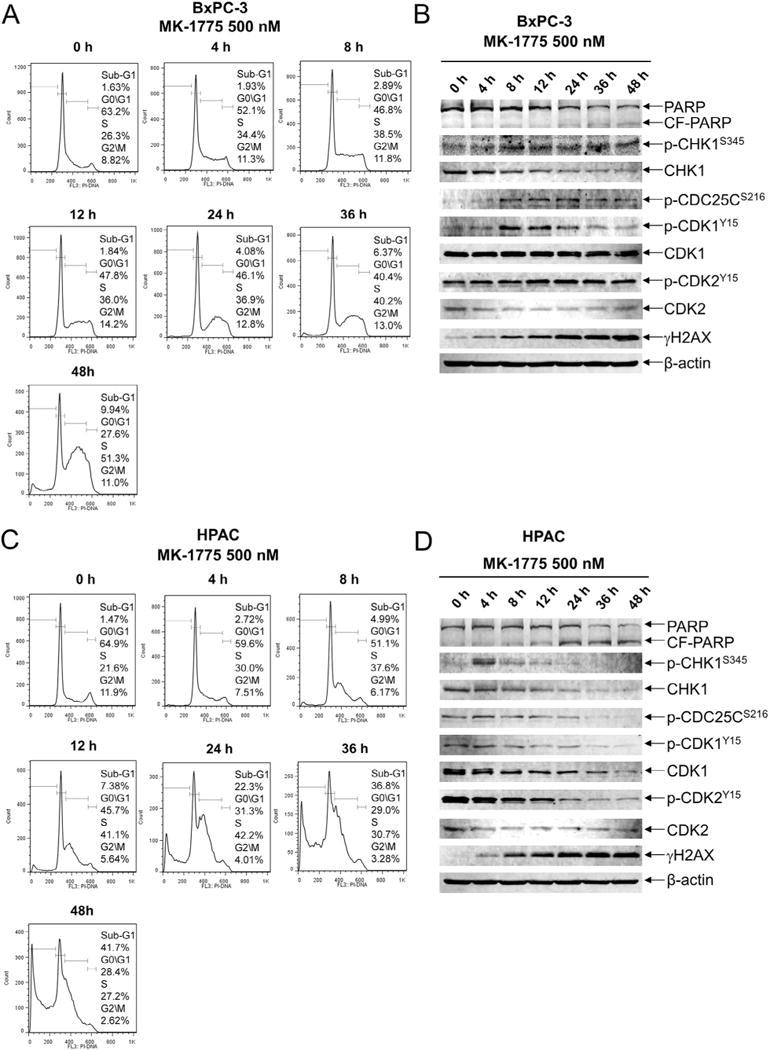
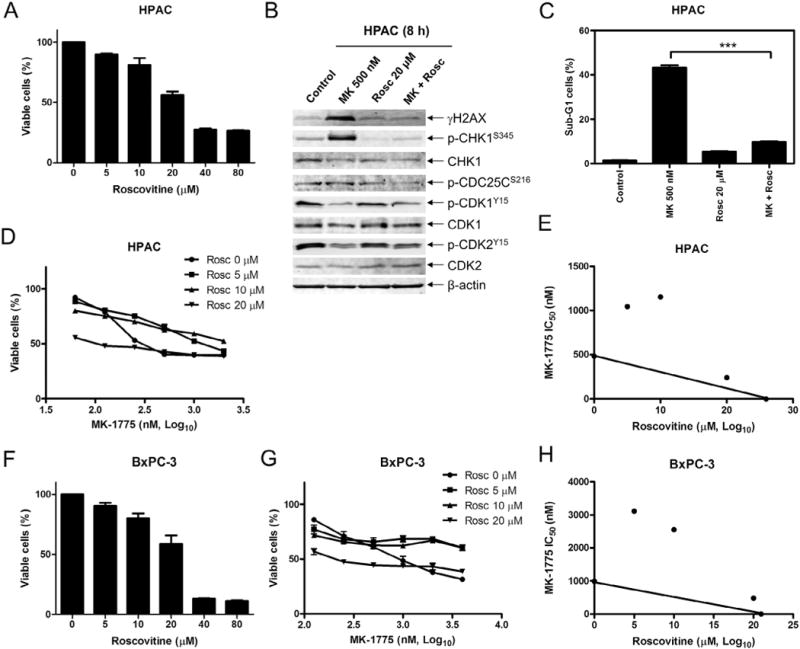
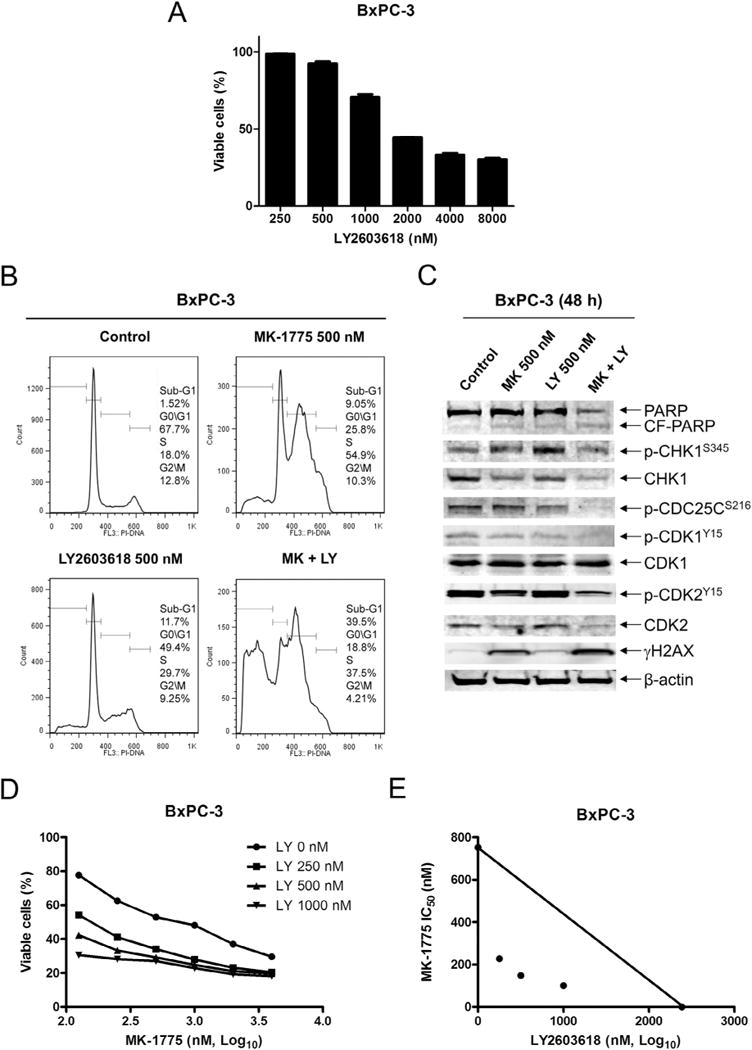
Similar articles
-
Targeting histone deacetylases (HDACs) and Wee1 for treating high-risk neuroblastoma.Pediatr Blood Cancer. 2015 Jan;62(1):52-9. doi: 10.1002/pbc.25232. Epub 2014 Oct 12. Pediatr Blood Cancer. 2015. PMID: 25308916
-
Synergistic anti-leukemic interactions between panobinostat and MK-1775 in acute myeloid leukemia ex vivo.Cancer Biol Ther. 2015;16(12):1784-93. doi: 10.1080/15384047.2015.1095406. Cancer Biol Ther. 2015. PMID: 26529495 Free PMC article.
-
Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma.Oncotarget. 2015 Feb 20;6(5):3394-408. doi: 10.18632/oncotarget.2583. Oncotarget. 2015. PMID: 25428911 Free PMC article.
-
Wee1 kinase as a target for cancer therapy.Cell Cycle. 2013 Oct 1;12(19):3159-64. doi: 10.4161/cc.26062. Epub 2013 Aug 26. Cell Cycle. 2013. PMID: 24013427 Free PMC article. Review.
-
Panobinostat as Pan-deacetylase Inhibitor for the Treatment of Pancreatic Cancer: Recent Progress and Future Prospects.Oncol Ther. 2016;4(1):73-89. doi: 10.1007/s40487-016-0023-1. Epub 2016 Jun 10. Oncol Ther. 2016. PMID: 28261641 Free PMC article. Review.
Cited by
-
Wee1 Inhibitor AZD1775 Combined with Cisplatin Potentiates Anticancer Activity against Gastric Cancer by Increasing DNA Damage and Cell Apoptosis.Biomed Res Int. 2018 Jun 7;2018:5813292. doi: 10.1155/2018/5813292. eCollection 2018. Biomed Res Int. 2018. PMID: 29977914 Free PMC article.
-
Low-Dose Radiation Enhanced Inhibition of Breast Tumor Xenograft and Reduced Myocardial Injury Induced by Doxorubicin.Dose Response. 2018 Dec 20;16(4):1559325818813061. doi: 10.1177/1559325818813061. eCollection 2018 Oct-Dec. Dose Response. 2018. PMID: 30622447 Free PMC article.
-
Chemogenetic profiling identifies RAD17 as synthetically lethal with checkpoint kinase inhibition.Oncotarget. 2015 Nov 3;6(34):35755-69. doi: 10.18632/oncotarget.5928. Oncotarget. 2015. PMID: 26437225 Free PMC article.
-
Suppression of Sirt1 sensitizes lung cancer cells to WEE1 inhibitor MK-1775-induced DNA damage and apoptosis.Oncogene. 2017 Dec 14;36(50):6863-6872. doi: 10.1038/onc.2017.297. Epub 2017 Sep 4. Oncogene. 2017. PMID: 28869605
-
Directing the use of DDR kinase inhibitors in cancer treatment.Expert Opin Investig Drugs. 2017 Dec;26(12):1341-1355. doi: 10.1080/13543784.2017.1389895. Epub 2017 Oct 14. Expert Opin Investig Drugs. 2017. PMID: 28984489 Free PMC article. Review.
References
-
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. - PubMed
-
- Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. - PubMed
-
- Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. - PubMed
-
- Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
