

p53 protein-mediated up-regulation of MAP kinase phosphatase 3 (MKP-3) contributes to the establishment of the cellular senescent phenotype through dephosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2)
- PMID: 25414256
- PMCID: PMC4294480
- DOI: 10.1074/jbc.M114.590943
p53 protein-mediated up-regulation of MAP kinase phosphatase 3 (MKP-3) contributes to the establishment of the cellular senescent phenotype through dephosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2)
Abstract
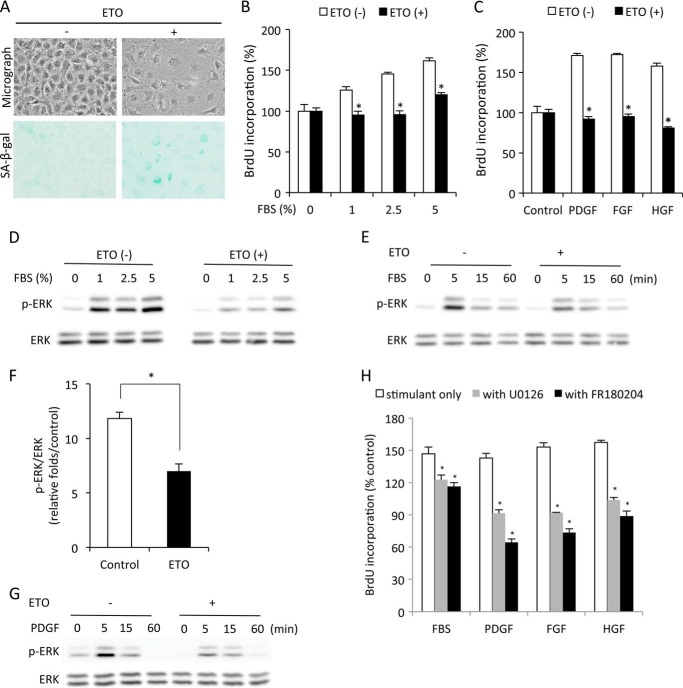
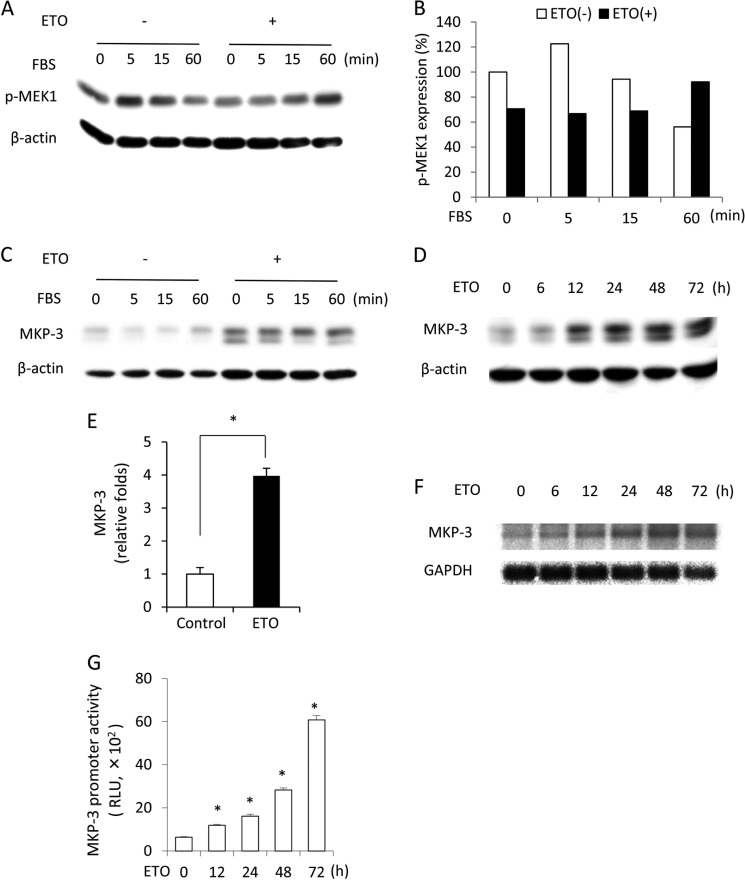
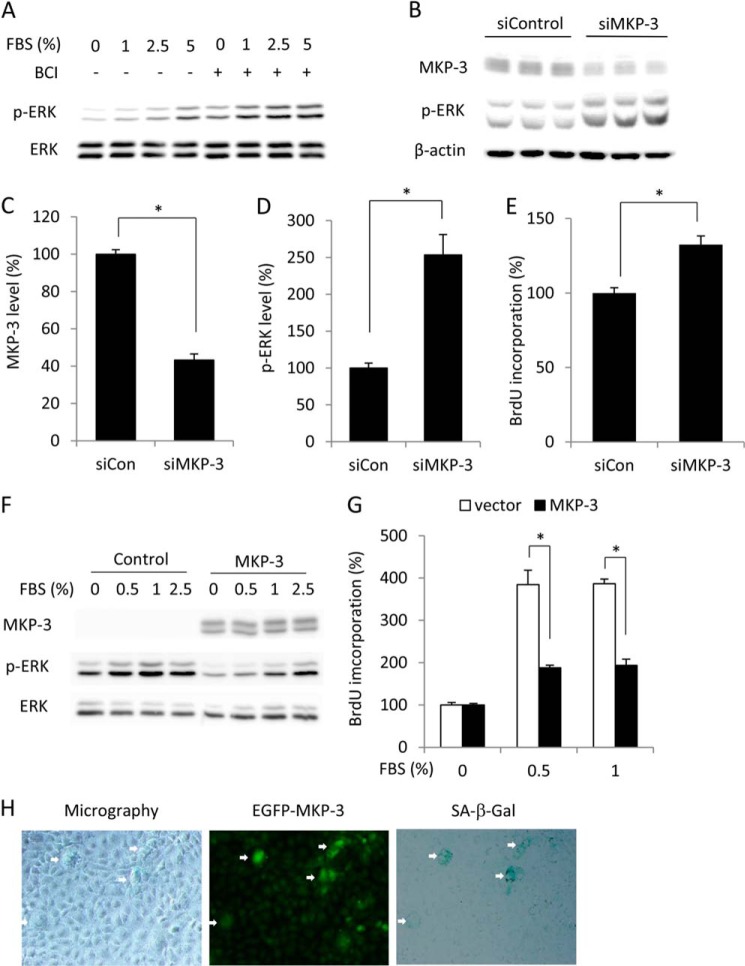
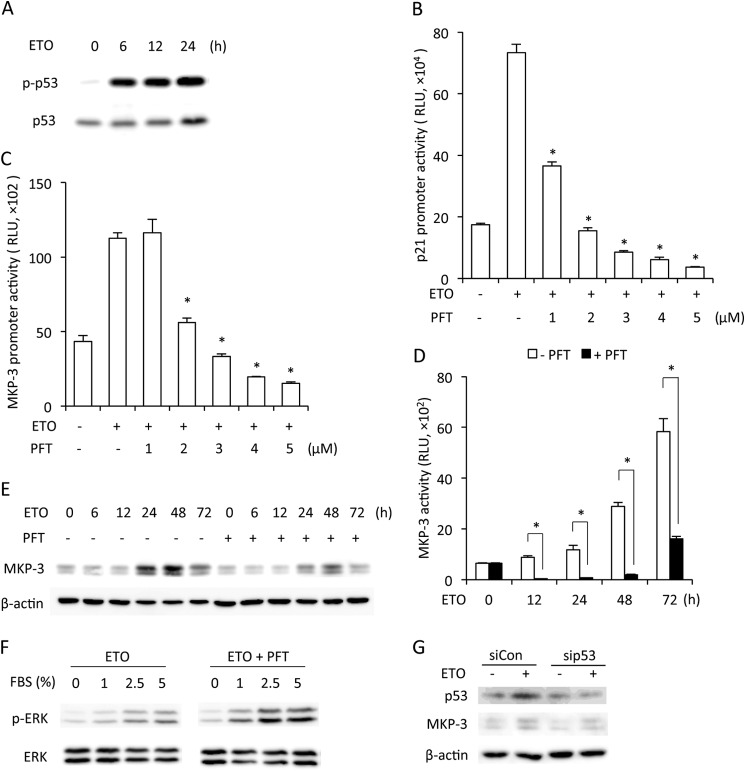
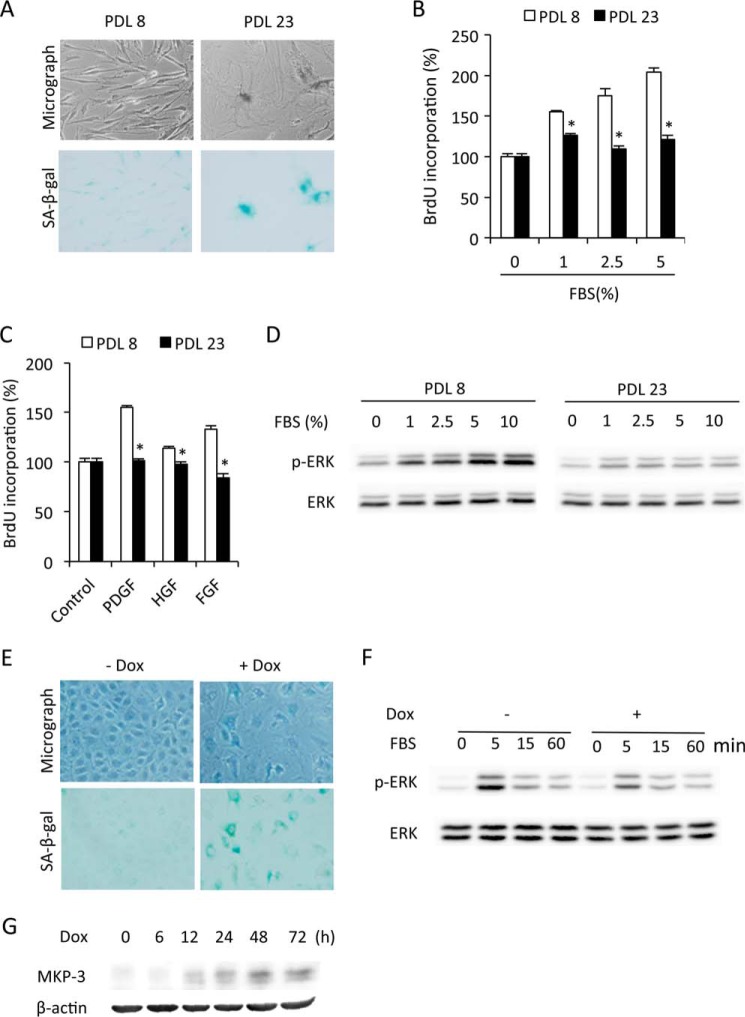
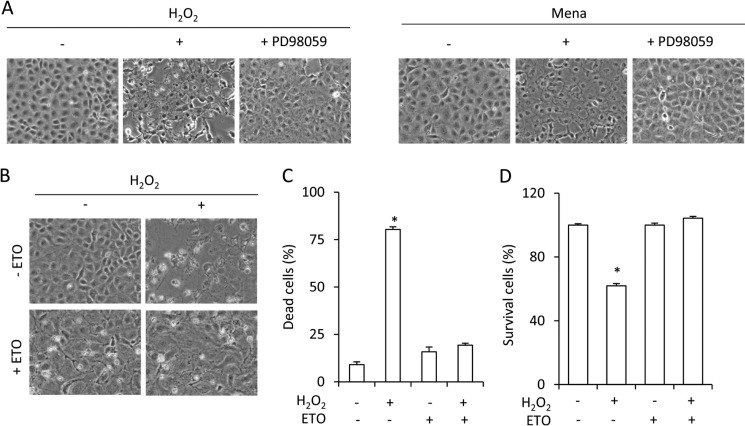
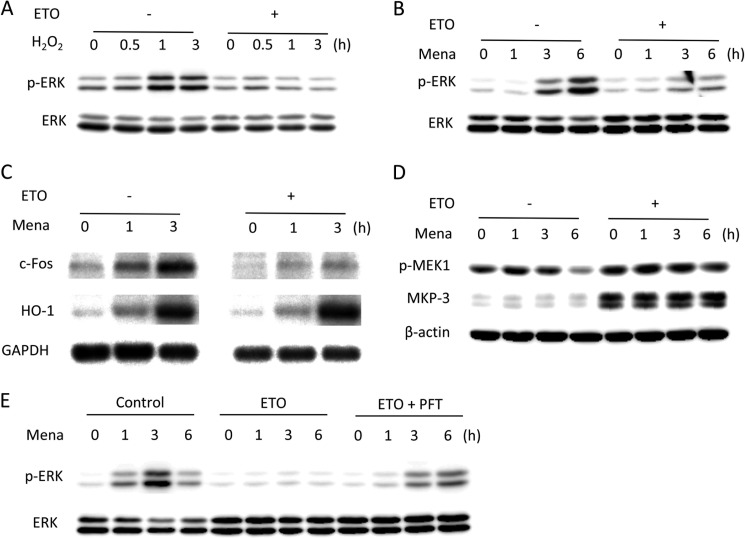
Growth arrest is one of the essential features of cellular senescence. At present, the precise mechanisms responsible for the establishment of the senescence-associated arrested phenotype are still incompletely understood. Given that ERK1/2 is one of the major kinases controlling cell growth and proliferation, we examined the possible implication of ERK1/2. Exposure of normal rat epithelial cells to etoposide caused cellular senescence, as manifested by enlarged cell size, a flattened cell body, reduced cell proliferation, enhanced β-galactosidase activity, and elevated p53 and p21. Senescent cells displayed a blunted response to growth factor-induced cell proliferation, which was preceded by impaired ERK1/2 activation. Further analysis revealed that senescent cells expressed a significantly higher level of mitogen-activated protein phosphatase 3 (MKP-3, a cytosolic ERK1/2-targeted phosphatase), which was suppressed by blocking the transcriptional activity of the tumor suppressor p53 with pifithrin-α. Inhibition of MKP-3 activity with a specific inhibitor or siRNA enhanced basal ERK1/2 phosphorylation and promoted cell proliferation. Apart from its role in growth arrest, impairment of ERK1/2 also contributed to the resistance of senescent cells to oxidant-elicited cell injury. These results therefore indicate that p53-mediated up-regulation of MKP-3 contributes to the establishment of the senescent cellular phenotype through dephosphorylating ERK1/2. Impairment of ERK1/2 activation could be an important mechanism by which p53 controls cellular senescence.
Keywords: Extracellular Signal-regulated Kinase (ERK); MAP Kinase Phosphatase 3 (MKP-3); Oxidative Stress; Proliferation; Senescence; p53.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
MAP kinase phosphatase-3 (MKP-3) is transcriptionally and post-translationally up-regulated by hCG and modulates cAMP-induced p21 expression in MA-10 Leydig cells.Mol Cell Endocrinol. 2013 May 22;371(1-2):174-81. doi: 10.1016/j.mce.2012.12.006. Epub 2012 Dec 20. Mol Cell Endocrinol. 2013. PMID: 23261984
-
ERK1/2-driven and MKP-mediated inhibition of EGF-induced ERK5 signaling in human proximal tubular cells.J Cell Physiol. 2007 Apr;211(1):88-100. doi: 10.1002/jcp.20909. J Cell Physiol. 2007. PMID: 17131384
-
Depletion of ERK2 but not ERK1 abrogates oncogenic Ras-induced senescence.Cell Signal. 2013 Dec;25(12):2540-7. doi: 10.1016/j.cellsig.2013.08.014. Epub 2013 Aug 30. Cell Signal. 2013. PMID: 23993963
-
Translational Control during Cellular Senescence.Mol Cell Biol. 2021 Jan 25;41(2):e00512-20. doi: 10.1128/MCB.00512-20. Print 2021 Jan 25. Mol Cell Biol. 2021. PMID: 33077499 Free PMC article. Review.
-
Mechanisms shaping the role of ERK1/2 in cellular senescence (Review).Mol Med Rep. 2019 Feb;19(2):759-770. doi: 10.3892/mmr.2018.9712. Epub 2018 Nov 29. Mol Med Rep. 2019. PMID: 30535440 Free PMC article. Review.
Cited by
-
MicroRNAs in the thyroid.Best Pract Res Clin Endocrinol Metab. 2016 Oct;30(5):603-619. doi: 10.1016/j.beem.2016.10.001. Epub 2016 Nov 1. Best Pract Res Clin Endocrinol Metab. 2016. PMID: 27923454 Free PMC article. Review.
-
Chinese 1 strain of Toxoplasma gondii excreted-secreted antigens negatively modulate Foxp3 via inhibition of the TGFßRII/Smad2/Smad3/Smad4 pathway.J Cell Mol Med. 2017 Sep;21(9):1944-1953. doi: 10.1111/jcmm.13115. Epub 2017 Mar 16. J Cell Mol Med. 2017. PMID: 28300338 Free PMC article.
-
ACTH(6-9)PGP Peptide Protects SH-SY5Y Cells from H2O2, tert-Butyl Hydroperoxide, and Cyanide Cytotoxicity via Stimulation of Proliferation and Induction of Prosurvival-Related Genes.Molecules. 2021 Mar 26;26(7):1878. doi: 10.3390/molecules26071878. Molecules. 2021. PMID: 33810344 Free PMC article.
-
miRNA and lncRNA Expression Networks Modulate Cell Cycle and DNA Repair Inhibition in Senescent Prostate Cells.Genes (Basel). 2022 Jan 24;13(2):208. doi: 10.3390/genes13020208. Genes (Basel). 2022. PMID: 35205253 Free PMC article.
-
Systematic screening of isogenic cancer cells identifies DUSP6 as context-specific synthetic lethal target in melanoma.Oncotarget. 2017 Apr 4;8(14):23760-23774. doi: 10.18632/oncotarget.15863. Oncotarget. 2017. PMID: 28423600 Free PMC article.
References
-
- Hayflick L. (1965) The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 37, 614–636 - PubMed
-
- Shay J. W., Roninson I. B. (2004) Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23, 2919–2933 - PubMed
-
- Shay J. W., Wright W. E. (2004) Telomeres are double-strand DNA breaks hidden from DNA damage responses. Mol. Cell 14, 420–421 - PubMed
-
- d'Adda di Fagagna F., Reaper P. M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N. P., Jackson S. P. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 - PubMed
-
- Herbig U., Jobling W. A., Chen B. P., Chen D. J., Sedivy J. M. (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
