

Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: involvement of CHOP through Wnt
- PMID: 25412307
- PMCID: PMC4260737
- DOI: 10.1038/cddis.2014.479
Activation of volume-sensitive outwardly rectifying chloride channel by ROS contributes to ER stress and cardiac contractile dysfunction: involvement of CHOP through Wnt
Abstract
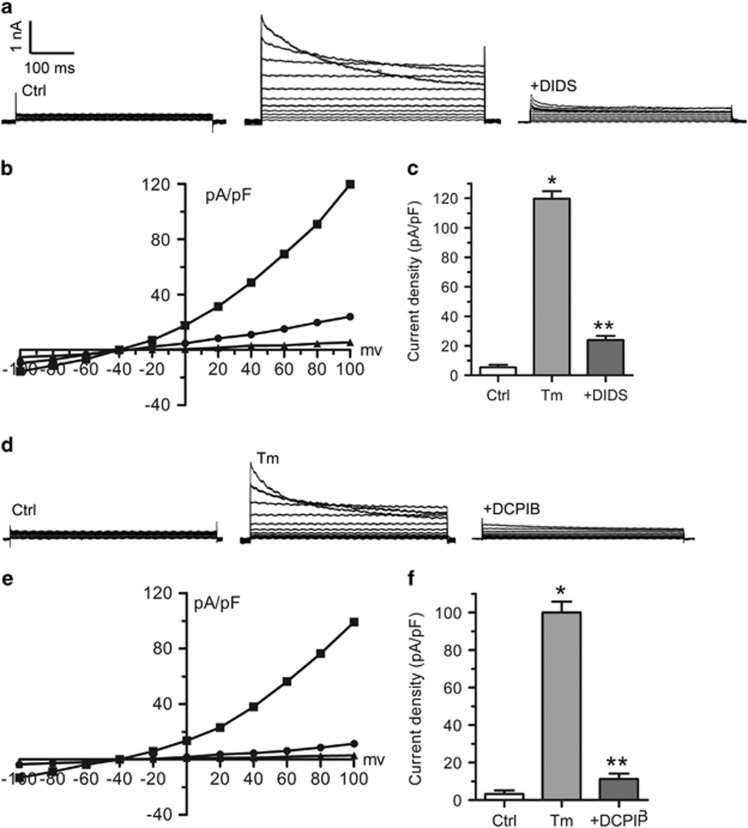
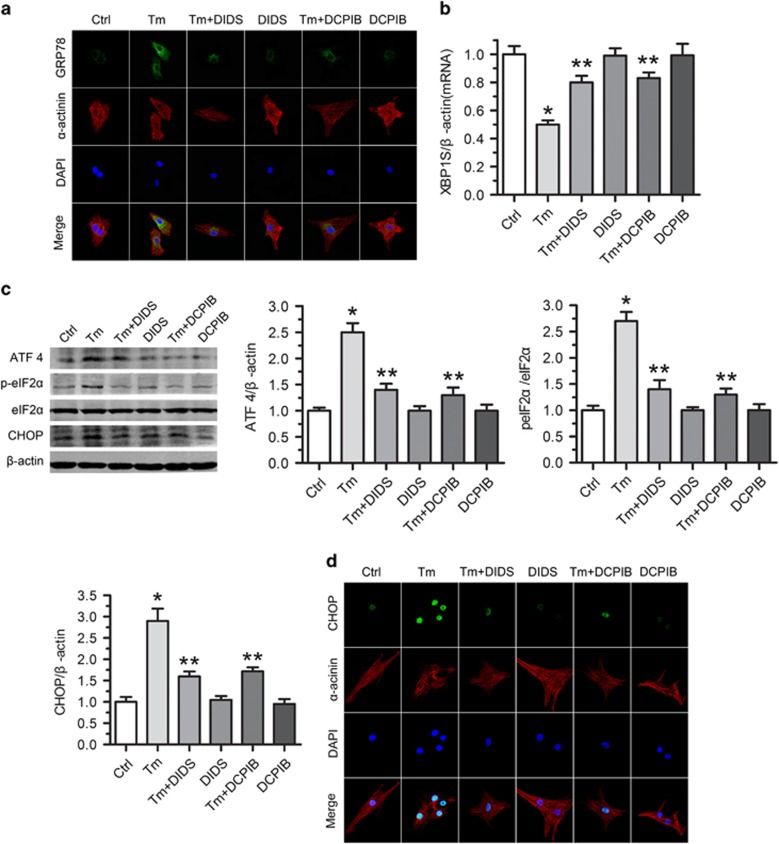
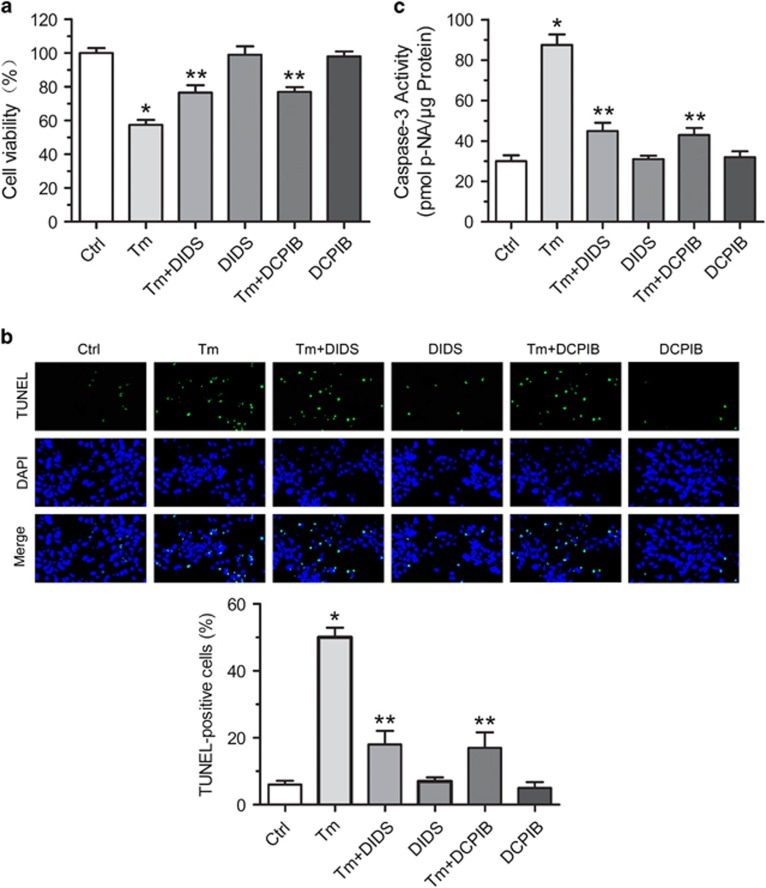
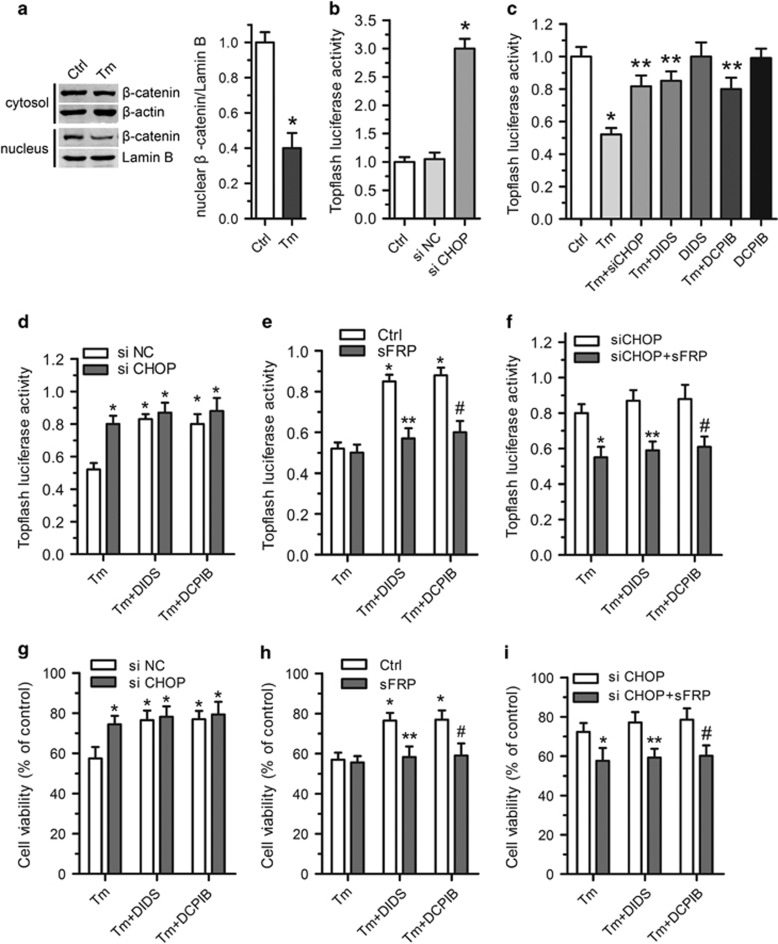
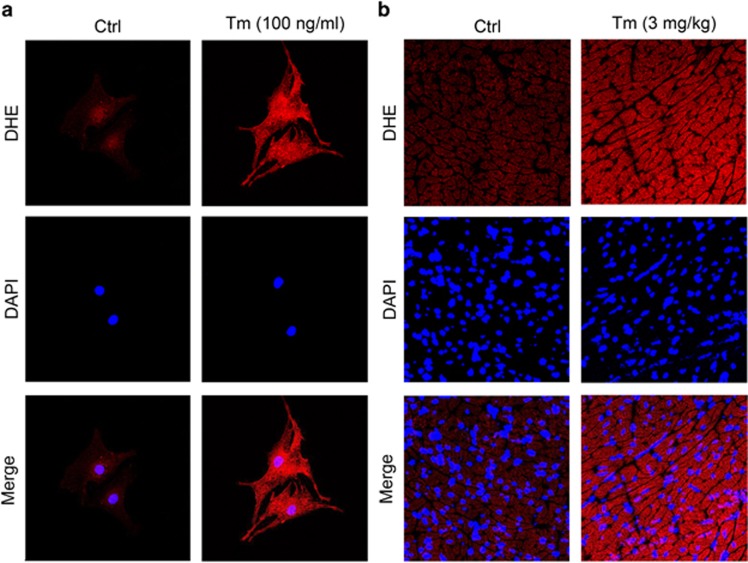
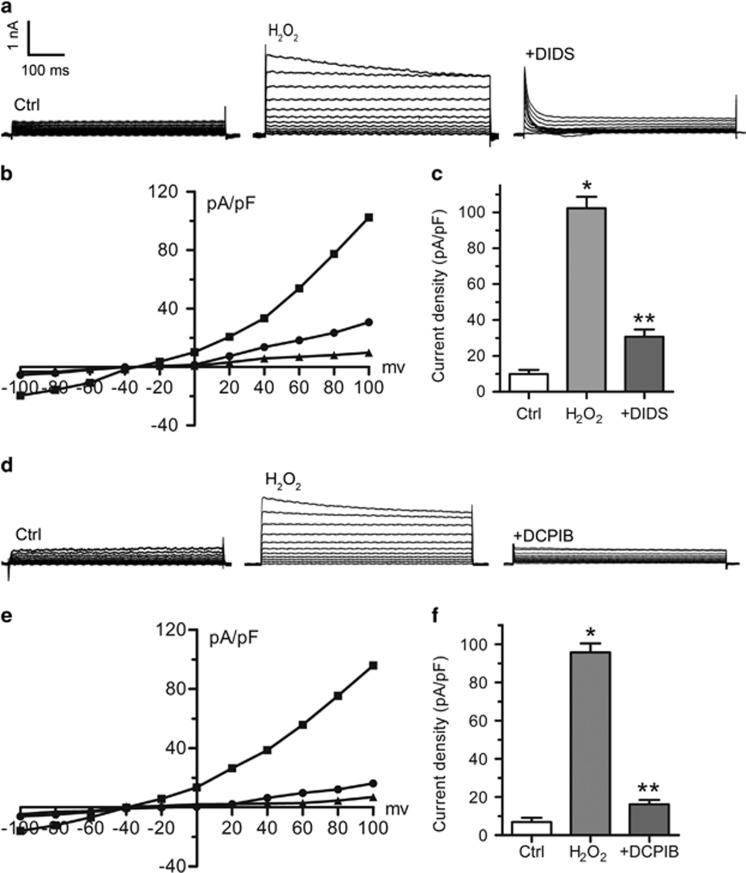
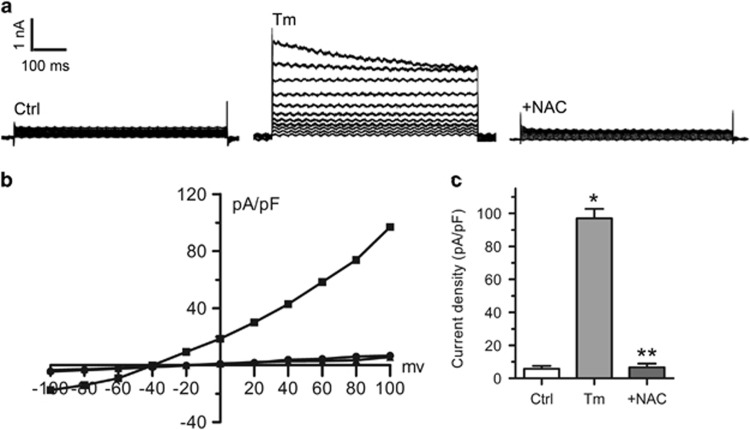
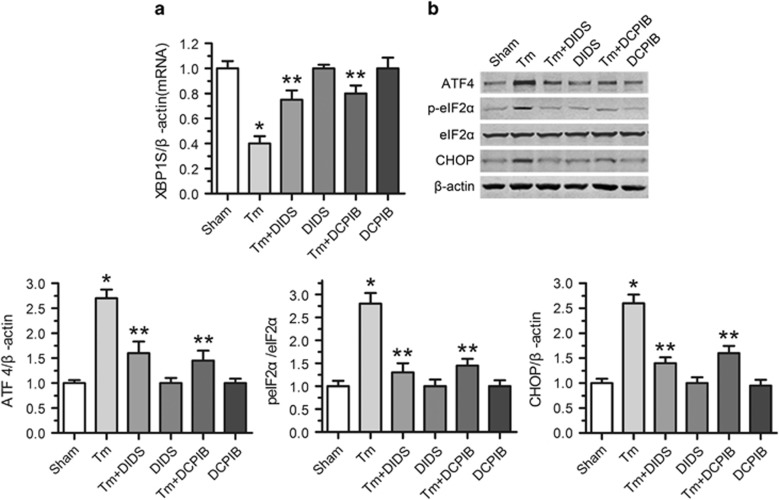
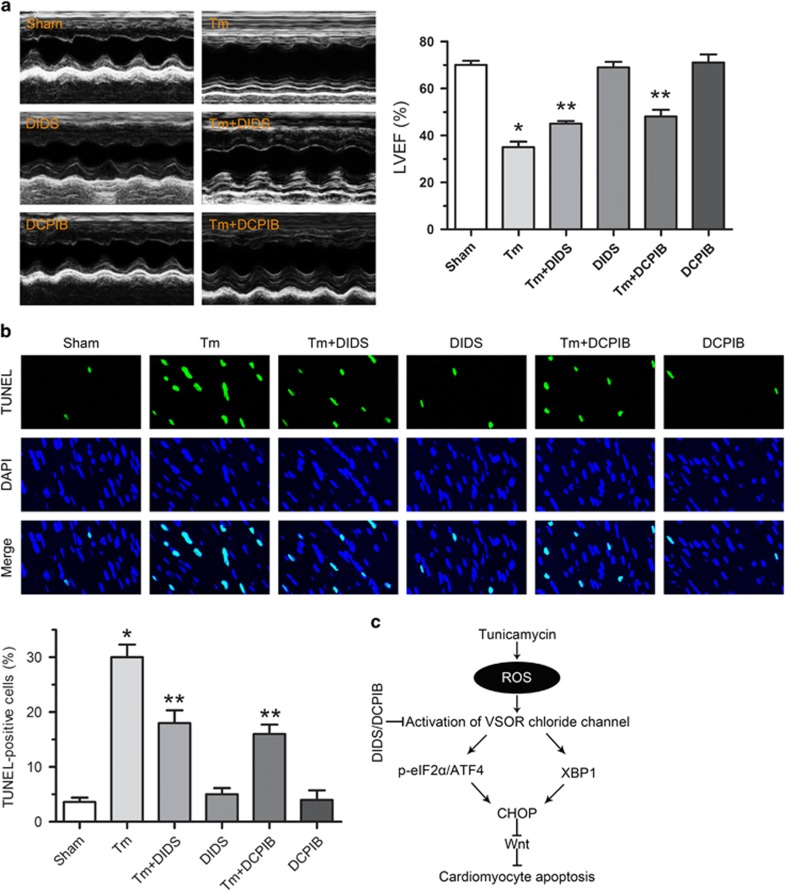
Endoplasmic reticulum (ER) stress occurring in stringent conditions is critically involved in cardiomyocytes apoptosis and cardiac contractile dysfunction (CCD). However, the molecular machinery that mediates cardiac ER stress and subsequent cell death remains to be fully deciphered, which will hopefully provide novel therapeutic targets for these disorders. Here, we establish tunicamycin-induced model of cardiomyocyte ER stress, which effectively mimicks pathological stimuli to trigger CCD. Tunicamycin activates volume-sensitive outward rectifying Cl(-) currents. Blockade of the volume-sensitive outwardly rectifying (VSOR) Cl(-) channel by 4,4'-diisothiocya-natostilbene-2,2'-disulfonic acid (DIDS), a non-selective Cl(-) channel blocker, and 4-(2-butyl-6,7-dichlor-2-cyclopentyl-indan-1-on-5-yl) oxybutyric acid (DCPIB), a selective VSOR Cl(-) channel blocker, improves cardiac contractility, which correlates with suppressed ER stress through inhibiting the canonical GRP78/eIF2α/ATF4 and XBP1 pathways, and promotes survival of cardiomyocytes by inverting tunicamycin-induced decrease of Wnt through the CHOP pathway. VSOR activation of tunicamycin-treated cardiomyocytes is attributed to increased intracellular levels of reactive oxygen species (ROS). Our study demonstrates a pivotal role of ROS/VSOR in mediating ER stress and functional impairment of cardiomyocytes via the CHOP-Wnt pathway, and suggests the therapeutic values of VSOR Cl(-) channel blockers against ER stress-associated cardiac anomalies.
Figures

Similar articles
-
Volume-sensitive outwardly rectifying chloride channel blockers protect against high glucose-induced apoptosis of cardiomyocytes via autophagy activation.Sci Rep. 2017 Mar 16;7:44265. doi: 10.1038/srep44265. Sci Rep. 2017. PMID: 28300155 Free PMC article.
-
Modulation of staurosporine-activated volume-sensitive outwardly rectifying Cl⁻ channel by PI3K/Akt in cardiomyocytes.Curr Pharm Des. 2013;19(27):4859-64. doi: 10.2174/1381612811319270008. Curr Pharm Des. 2013. PMID: 23323619
-
Gambogenic acid triggers apoptosis in human nasopharyngeal carcinoma CNE-2Z cells by activating volume-sensitive outwardly rectifying chloride channel.Fitoterapia. 2019 Mar;133:150-158. doi: 10.1016/j.fitote.2019.01.002. Epub 2019 Jan 14. Fitoterapia. 2019. PMID: 30654125
-
Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death.J Membr Biol. 2006 Jan;209(1):21-9. doi: 10.1007/s00232-005-0836-6. Epub 2006 Apr 17. J Membr Biol. 2006. PMID: 16685598 Review.
-
The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress.Curr Mol Med. 2016;16(6):533-44. doi: 10.2174/1566524016666160523143937. Curr Mol Med. 2016. PMID: 27211800 Free PMC article. Review.
Cited by
-
Overexpression and Activation of αvβ3 Integrin Differentially Affects TGFβ2 Signaling in Human Trabecular Meshwork Cells.Cells. 2021 Jul 29;10(8):1923. doi: 10.3390/cells10081923. Cells. 2021. PMID: 34440692 Free PMC article.
-
Volume-sensitive outwardly rectifying chloride channel blockers protect against high glucose-induced apoptosis of cardiomyocytes via autophagy activation.Sci Rep. 2017 Mar 16;7:44265. doi: 10.1038/srep44265. Sci Rep. 2017. PMID: 28300155 Free PMC article.
-
Pathway analysis identifies altered mitochondrial metabolism, neurotransmission, structural pathways and complement cascade in retina/RPE/ choroid in chick model of form-deprivation myopia.PeerJ. 2018 Jun 27;6:e5048. doi: 10.7717/peerj.5048. eCollection 2018. PeerJ. 2018. PMID: 29967729 Free PMC article.
-
Long non-coding RNA MEG3 knockdown attenuates endoplasmic reticulum stress-mediated apoptosis by targeting p53 following myocardial infarction.J Cell Mol Med. 2019 Dec;23(12):8369-8380. doi: 10.1111/jcmm.14714. Epub 2019 Oct 20. J Cell Mol Med. 2019. PMID: 31631486 Free PMC article.
-
TBHQ Attenuates Neurotoxicity Induced by Methamphetamine in the VTA through the Nrf2/HO-1 and PI3K/AKT Signaling Pathways.Oxid Med Cell Longev. 2020 Apr 13;2020:8787156. doi: 10.1155/2020/8787156. eCollection 2020. Oxid Med Cell Longev. 2020. PMID: 32351675 Free PMC article.
References
-
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. - PubMed
-
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. - PubMed
-
- He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
