

Review
doi: 10.1038/nrg3849.
Epub 2014 Nov 18.
Cystic fibrosis genetics: from molecular understanding to clinical application
Affiliations
- PMID: 25404111
- PMCID: PMC4364438
- DOI: 10.1038/nrg3849
Item in Clipboard
Review
Cystic fibrosis genetics: from molecular understanding to clinical application
Nat Rev Genet.
2015 Jan.
Abstract
The availability of the human genome sequence and tools for interrogating individual genomes provide an unprecedented opportunity to apply genetics to medicine. Mendelian conditions, which are caused by dysfunction of a single gene, offer powerful examples that illustrate how genetics can provide insights into disease. Cystic fibrosis, one of the more common lethal autosomal recessive Mendelian disorders, is presented here as an example. Recent progress in elucidating disease mechanism and causes of phenotypic variation, as well as in the development of treatments, demonstrates that genetics continues to play an important part in cystic fibrosis research 25 years after the discovery of the disease-causing gene.
Figures
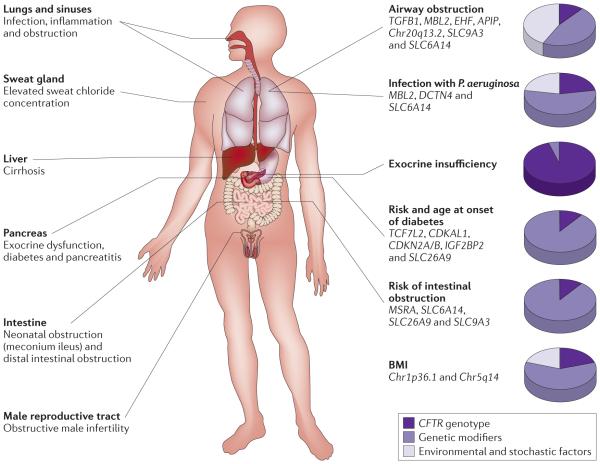
A diagnosis of cystic fibrosis is based on the presence of clinical findings shown on the left, along with an elevated sweat chloride concentration (>60mM). The degree of organ system dysfunction varies considerably among affected individuals. Genetic modifiers and non-genetic factors both contribute to airwayobstruction and infection with Pseudomonas aeruginosa -two traits that define lung disease in cystic fibrosis. Cystic fibrosis transmembrane conductance regulator (CFTR) genotype is the primarydeterm inant of the degree of pancreatic exocrine dysfunction. The presence of CFTR variants associated with severe pancreatic exocrine dysfunction is essentially a pre-requisite for the development of diabetes and intestinal obstruction. In the setting of severe endocrine dysfunction, genetic modifiers determine when, and if, diabetes occurs and whether neonatal intestinal obstruction occurs. Genetic variation plays the predominant part in nutritional status as assessed by body mass index (BMI)70.
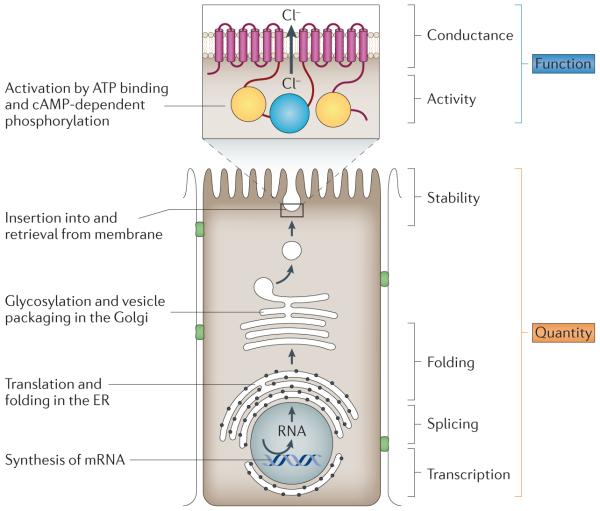
Thedegree to which epithelial ion transport is altered in an individual with cystic fibrosis is determined by the effect of each disease-causing variant on the quantity and the function of cystic fibrosis transmembrane conductance regulator (CFTR). The key steps of CFTR biogenesis in an epithelial cell are depicted. The membrane-spanning domains of CFTR are shown as red boxes, the two nucleotide-binding domains as yellow circles, and the regulatory domain as a blue circle. The quantity of CFTRprotein in the apical cell membrane is a product of the amount of RNA transcribed, the efficiencyof RNA splicing, the fraction of protein correctly folded and the stability of the protein in the membrane. The level and/or content of CFTR transcripts can be affected by disease-causing variants in the promoter (for example, c.-234T→A (also known as -102T→A in legacy nomenclature)) and splice sites (for example, c. 3717+12191 C-→T (legacy 3849+10 kb C→1)), or by variants that introduce a premature termination codon (PTC) and that lead to RNA decay (for example, p.Gly542X; (legacy G542X). The processing of CFTR can be altered by variants that cause aberrant folding of the protein, leading to degradation (for example, p.PheS08del (legacy F508del)), or by variants that cause reduced membrane stability as a result of increased rates of endocytosis (for example, p.Asn287Tyr (legacy N287Y)). The function of C FTR is dependent on activity of the ion channel and on the efficiency of conductance of ions through the channel. Disease-causing variants cause reduction in activity(for example, p.Gly551Asp (legacy G551D) or changes in the conduct ion proper!ies of the chloride channel (for example, p.Arg334Trp (legacyR334W)). cAMP,cyclicAMP; ER, endoplasmic reticulum.
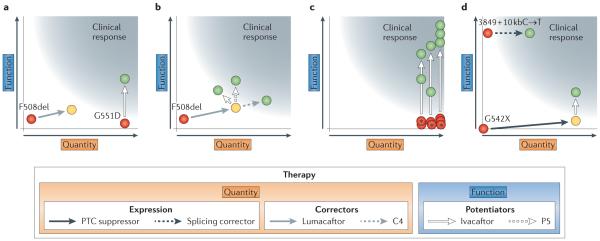
The effects of various therapies (shown as arrows) on the quantity and/or function of mutant cystic fibrosis transmembrane conductance regulator (C FTR; FIG. 2) are shown. At least 10% of 'normal' CFTR function is required for 'clinical response' in lung function (grey region). The indistinct border of the clinical response accommodates various estimates of the amount of C FTR ion transport that have to be achieved and the probability that the product of quantity and function may generate a nonlinear function in certain circumstances. Red circles indicate the approximate product of quantity and function of CFTR bearing the indicated variant. Yellow circles indicate a shift in the product of quantity and function that does not achieve a clinical response, and green circles indicate a shift that does produce a clinical response. a I Single-drug strategies for G551D (also known as p. Gly551Asp) and F508del (also known as p.Phe508del) are shown. CFTR bearing the G551Dvariant is found at normal levels in the cell membrane but cannot be activated. The potent iator ivacaftor increases the activity of G551D-C FTR, thereby increasing chloride transport in airway epithelia to a level that produces a clinical response in the lungs. The common variant F508del causes a defect in protein folding, which greatlyd iminishes CFTR quantity at the plasma membrane and reduces both membrane residency and channel activity. Application of lumacaftor to cells increases the quantity and, to a lesser degree, the chloride transport of F508del-CFTR, but not to a level that produces a clinical response in the lungs. b ICombinatorial strategies for F508del are shown. In cell-based studies, chronic administration of ivacaftor counteracts the increase in CFTR stability conferred by lumacaftor,. Use of other potentiators that do not antagonize the effect oflumacaftor, such as the investigational compound P5, could provide higher therapeutic benefit. An alternative approach is to combine correctors that affect different stages of CFTR folding, such as lumacaftor and C4 (REF. 32). c ITheratype strategy for rare variants is shown. Red circles with asterisks ind icatevar iants that permit product ion of normal or near-normal quantity of CFTR but that alter the activity of the C FTR chloride channel. As this functional defect is similar to that caused by G551D, each variant is tested for response to ivacaftor in cell-based studies. Recovery of C FTR function that exceeds 10% of levels seen in normal individuals indicates that a clinical response is expected, thereby justifying clinical trials. dIStrategies for nonsense and splice site variants are shown. The splice sitevariant 3849+ 10kbC→T (also known as c.3717+12191C→1) activates a cryptic splice donor site, causing reduction of the full-length CFTR transcript to −8% of normal levels. Suppression of the cryptic splice site can increase CFTR protein levels above 10%of normal. The nonsense variant G542X (also known as p.Gly542X) introduces a premature termination codon (PTC) that causes severe reduction in mRNA levels and an absence of the CFTRprotein. Use of PTC suppressors increases transcript and protein levels, leading to a modest recovery of CFTR function that falls short of a din ical response. Combining ivacaftor with a PTC suppressor produces a 2. 5--4-fold increase in function of G542X-CFTR that could be sufficient to produce a clinical response in lung function.
Similar articles
-
[Modifier genes and cystic fibrosis].Arch Pediatr. 2006 Jan;13(1):57-63. doi: 10.1016/j.arcped.2005.09.029. Epub 2005 Nov 7. Arch Pediatr. 2006. PMID: 16274977 Review. French.
-
Molecular Diagnosis of Cystic Fibrosis.Curr Protoc Hum Genet. 2016 Jan 1;88:9.28.1-9.28.6. doi: 10.1002/0471142905.hg0928s88. Curr Protoc Hum Genet. 2016. PMID: 26724724
-
Precision Genomic Medicine in Cystic Fibrosis.Clin Transl Sci. 2015 Oct;8(5):606-10. doi: 10.1111/cts.12292. Epub 2015 Jun 15. Clin Transl Sci. 2015. PMID: 26073768 Free PMC article. Review.
-
First DNA-based test to detect cystic fibrosis.FDA Consum. 2005 Jul-Aug;39(4):4. FDA Consum. 2005. PMID: 16252390 No abstract available.
-
Molecular diagnosis of cystic fibrosis.Expert Rev Mol Diagn. 2002 May;2(3):240-56. doi: 10.1586/14737159.2.3.240. Expert Rev Mol Diagn. 2002. PMID: 12050863 Review.
Cited by
-
Deep representation learning of electronic health records to unlock patient stratification at scale.NPJ Digit Med. 2020 Jul 17;3:96. doi: 10.1038/s41746-020-0301-z. eCollection 2020. NPJ Digit Med. 2020. PMID: 32699826 Free PMC article.
-
Autophagy in Pulmonary Diseases.Am J Respir Crit Care Med. 2016 Nov 15;194(10):1196-1207. doi: 10.1164/rccm.201512-2468SO. Am J Respir Crit Care Med. 2016. PMID: 27579514 Free PMC article. Review.
-
Chronic rhinosinusitis in patients with cystic fibrosis-Current management and new treatments.Laryngoscope Investig Otolaryngol. 2020 Jun 13;5(3):368-374. doi: 10.1002/lio2.401. eCollection 2020 Jun. Laryngoscope Investig Otolaryngol. 2020. PMID: 32596478 Free PMC article. Review.
-
Influenza, SARS-CoV-2, and Their Impact on Chronic Lung Diseases and Fibrosis: Exploring Therapeutic Options.Am J Pathol. 2024 Oct;194(10):1807-1822. doi: 10.1016/j.ajpath.2024.06.004. Epub 2024 Jul 18. Am J Pathol. 2024. PMID: 39032604 Review.
-
Antibacterial Activity of a Natural Clay Mineral against Burkholderia cepacia Complex and Other Bacterial Pathogens Isolated from People with Cystic Fibrosis.Microorganisms. 2023 Jan 6;11(1):150. doi: 10.3390/microorganisms11010150. Microorganisms. 2023. PMID: 36677442 Free PMC article.
References
-
- Cystic Fibrosis Foundation . Cystic Fibrosis Foundation Patient Registry Annual Data Report 2011. Cystic Fibrosis Foundation; 2012.
-
- Rommens JM, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. - PubMed
-
- Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Kerem B, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. References 3-5 are landmark papers from 25 years ago reporting the discovery of the CFTR gene. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
