

PUL21a-Cyclin A2 interaction is required to protect human cytomegalovirus-infected cells from the deleterious consequences of mitotic entry
- PMID: 25393019
- PMCID: PMC4231158
- DOI: 10.1371/journal.ppat.1004514
PUL21a-Cyclin A2 interaction is required to protect human cytomegalovirus-infected cells from the deleterious consequences of mitotic entry
Abstract
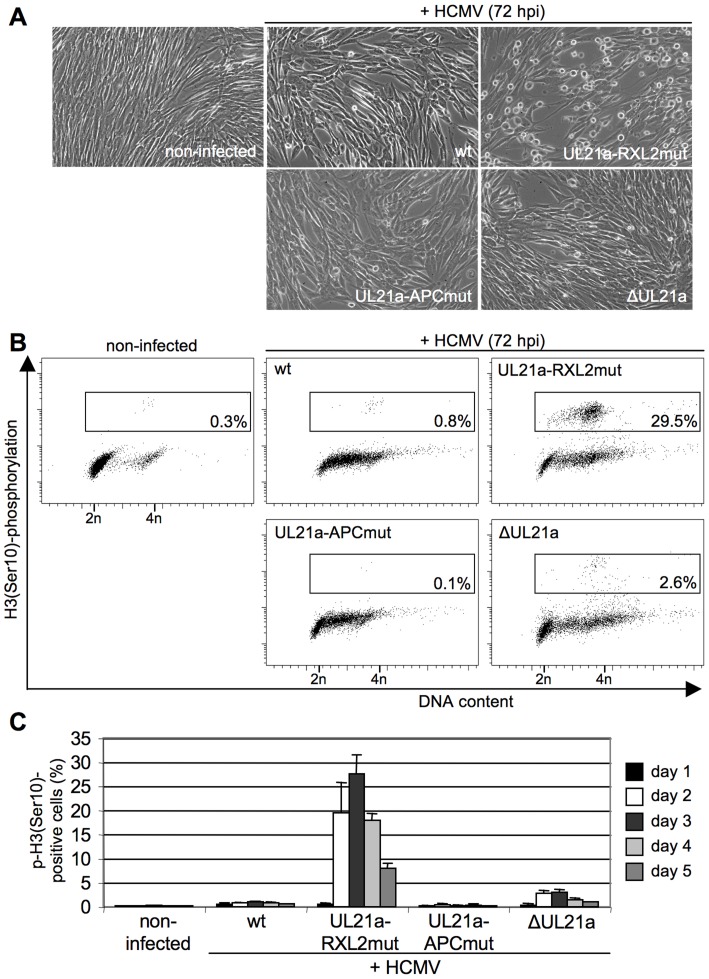
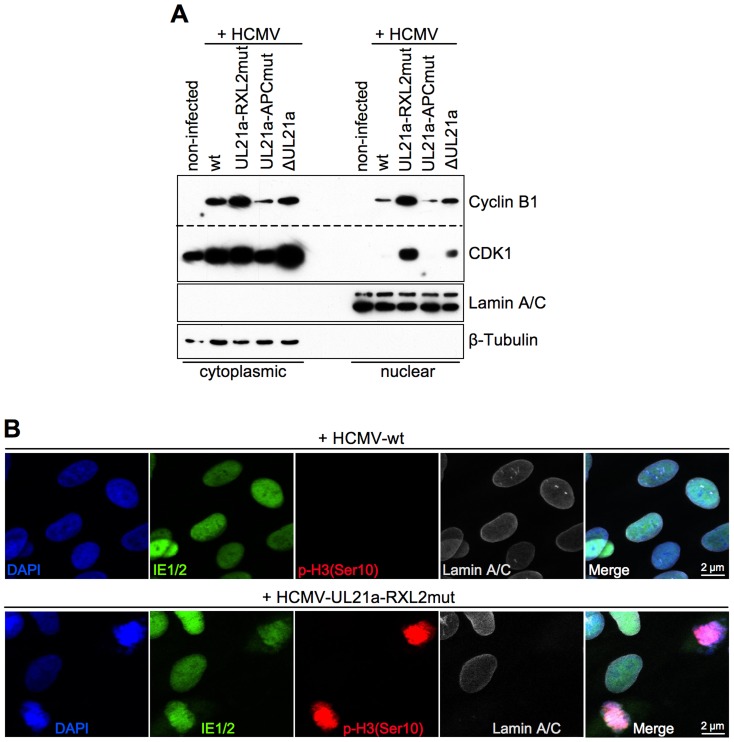
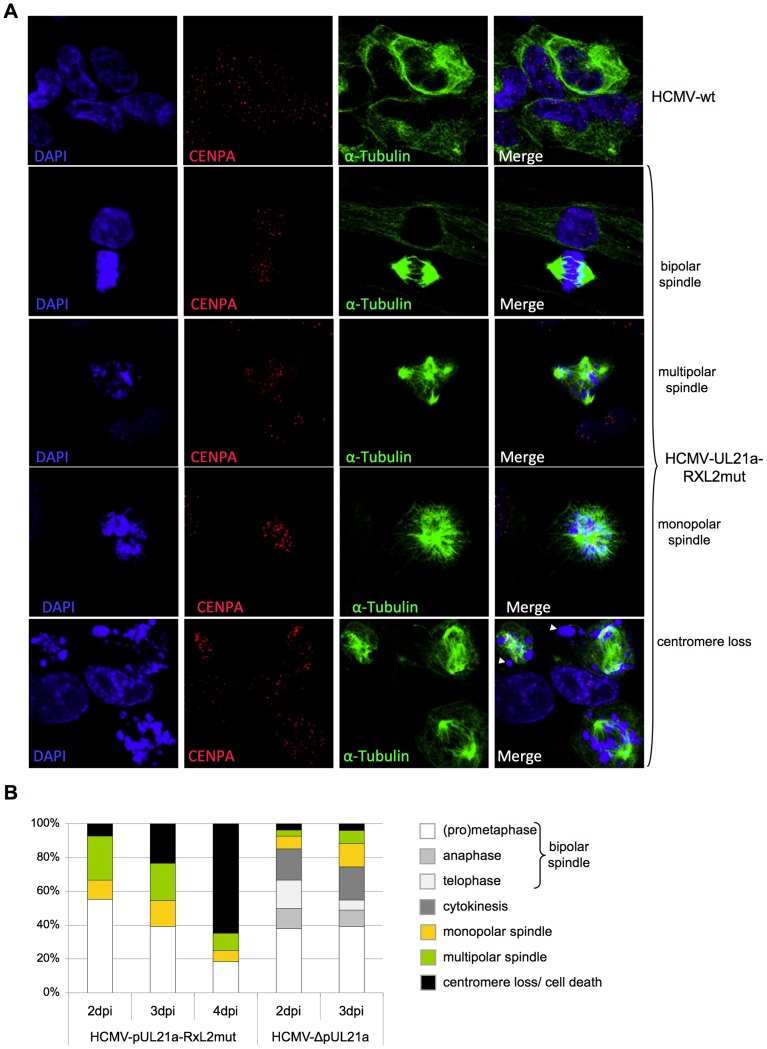
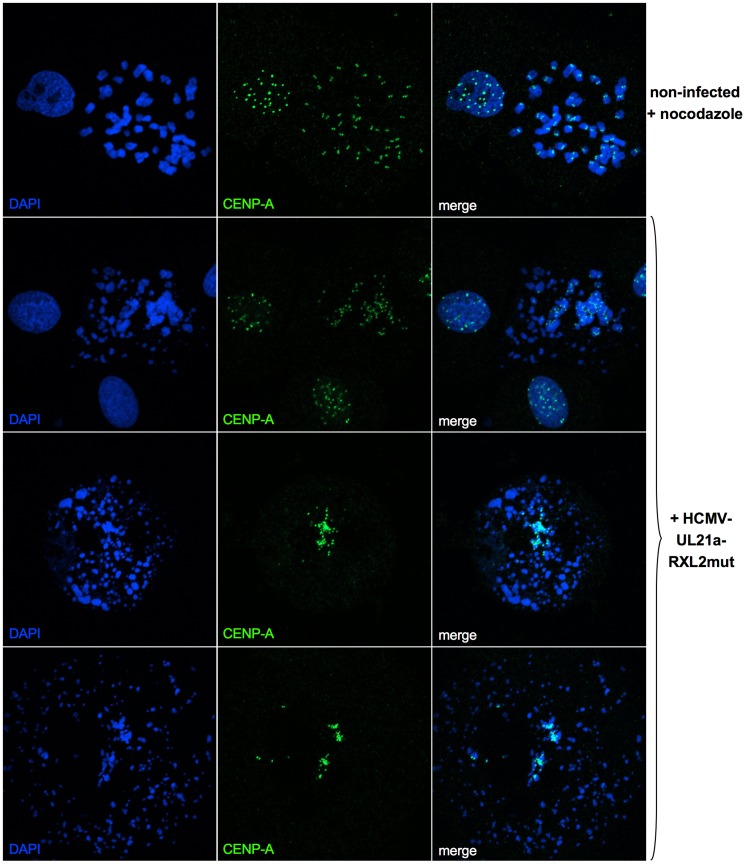
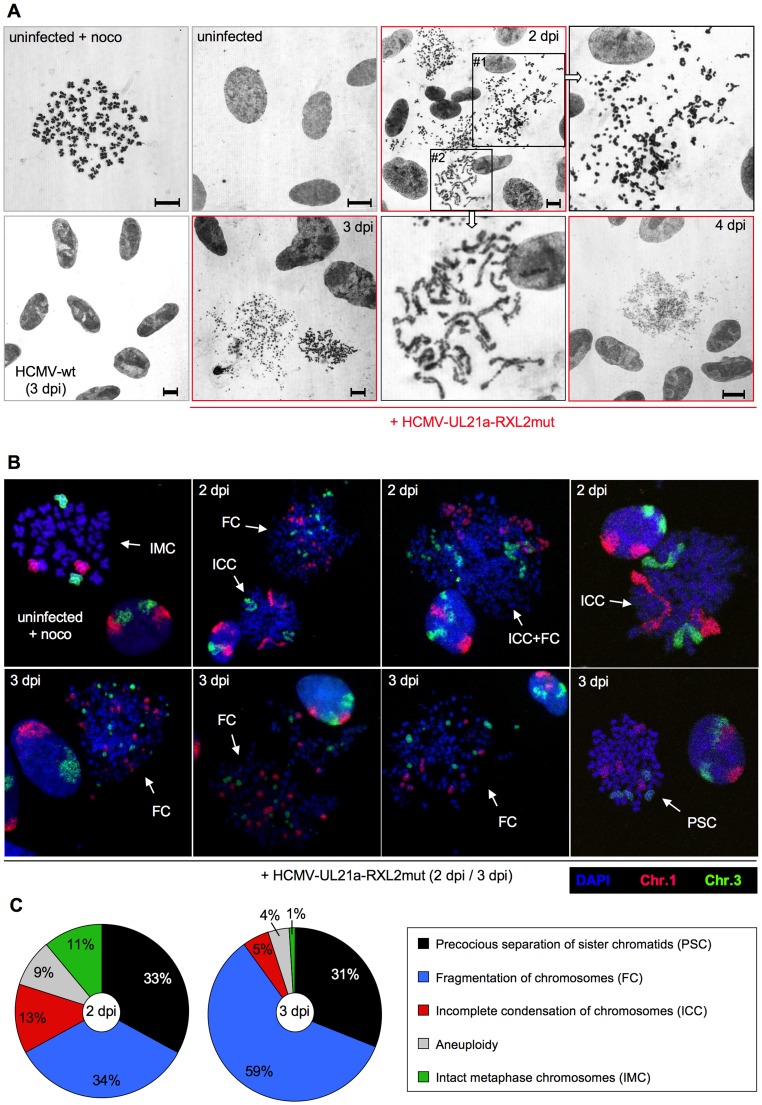
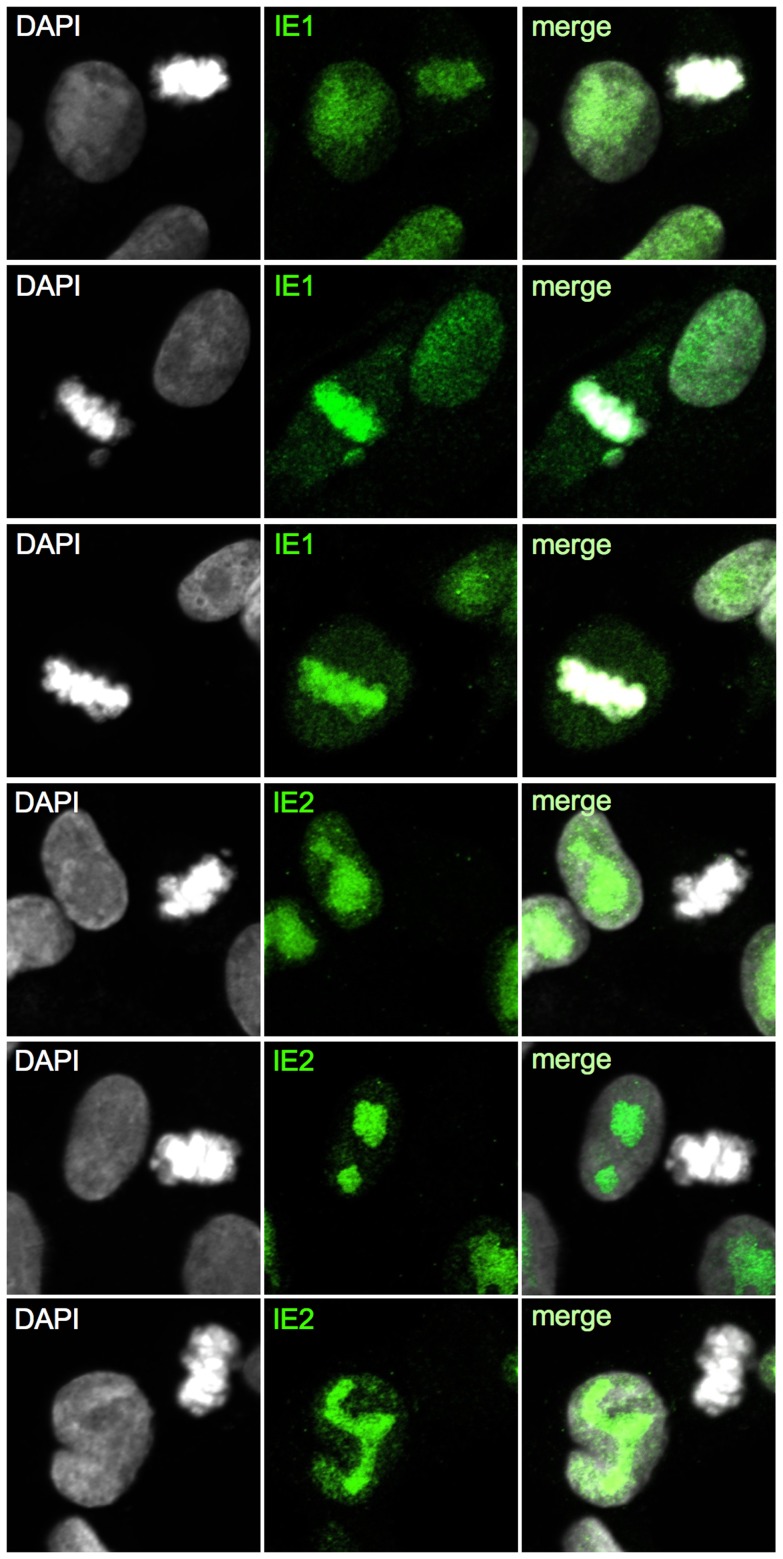
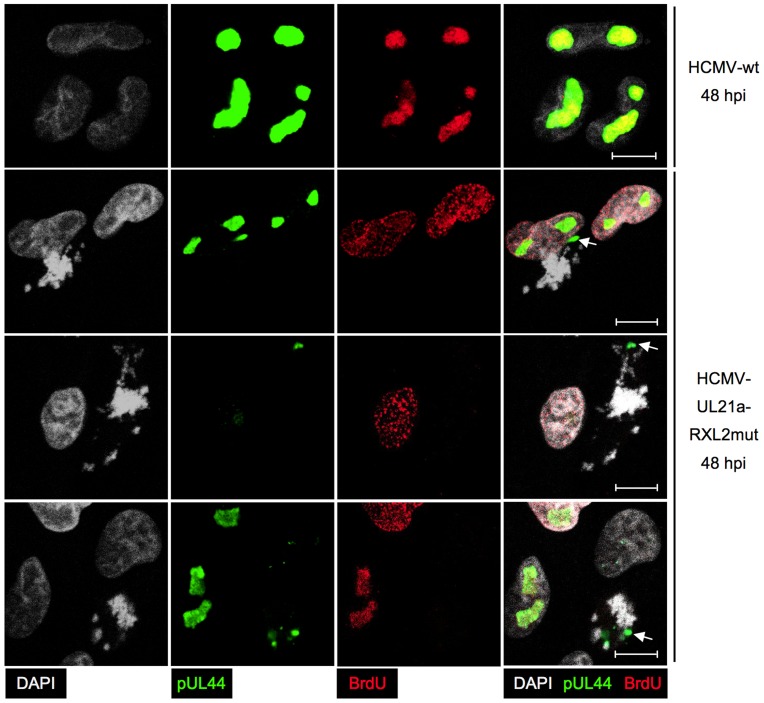
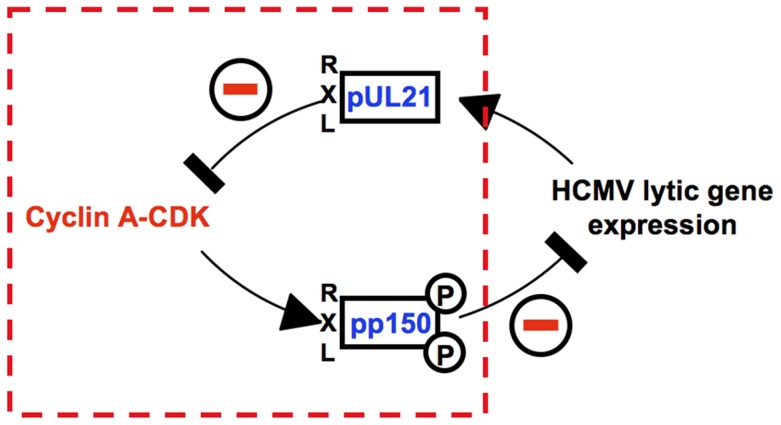
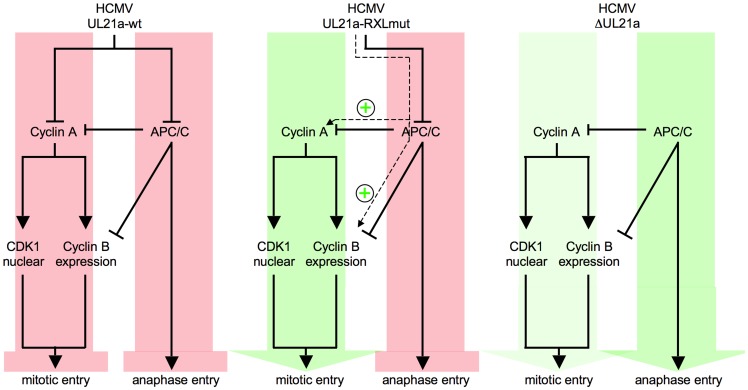
Entry into mitosis is accompanied by dramatic changes in cellular architecture, metabolism and gene expression. Many viruses have evolved cell cycle arrest strategies to prevent mitotic entry, presumably to ensure sustained, uninterrupted viral replication. Here we show for human cytomegalovirus (HCMV) what happens if the viral cell cycle arrest mechanism is disabled and cells engaged in viral replication enter into unscheduled mitosis. We made use of an HCMV mutant that, due to a defective Cyclin A2 binding motif in its UL21a gene product (pUL21a), has lost its ability to down-regulate Cyclin A2 and, therefore, to arrest cells at the G1/S transition. Cyclin A2 up-regulation in infected cells not only triggered the onset of cellular DNA synthesis, but also promoted the accumulation and nuclear translocation of Cyclin B1-CDK1, premature chromatin condensation and mitotic entry. The infected cells were able to enter metaphase as shown by nuclear lamina disassembly and, often irregular, metaphase spindle formation. However, anaphase onset was blocked by the still intact anaphase promoting complex/cyclosome (APC/C) inhibitory function of pUL21a. Remarkably, the essential viral IE2, but not the related chromosome-associated IE1 protein, disappeared upon mitotic entry, suggesting an inherent instability of IE2 under mitotic conditions. Viral DNA synthesis was impaired in mitosis, as demonstrated by the abnormal morphology and strongly reduced BrdU incorporation rates of viral replication compartments. The prolonged metaphase arrest in infected cells coincided with precocious sister chromatid separation and progressive fragmentation of the chromosomal material. We conclude that the Cyclin A2-binding function of pUL21a contributes to the maintenance of a cell cycle state conducive for the completion of the HCMV replication cycle. Unscheduled mitotic entry during the course of the HCMV replication has fatal consequences, leading to abortive infection and cell death.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Human cytomegalovirus riding the cell cycle.Med Microbiol Immunol. 2015 Jun;204(3):409-19. doi: 10.1007/s00430-015-0396-z. Epub 2015 Mar 17. Med Microbiol Immunol. 2015. PMID: 25776080 Review.
-
Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection.Virology. 2000 Aug 15;274(1):39-55. doi: 10.1006/viro.2000.0448. Virology. 2000. PMID: 10936087
-
Inhibition of human cytomegalovirus immediate-early gene expression by cyclin A2-dependent kinase activity.J Virol. 2012 Sep;86(17):9369-83. doi: 10.1128/JVI.07181-11. Epub 2012 Jun 20. J Virol. 2012. PMID: 22718829 Free PMC article.
-
Repression of HMGA2 gene expression by human cytomegalovirus involves the IE2 86-kilodalton protein and is necessary for efficient viral replication and inhibition of cyclin A transcription.J Virol. 2006 Oct;80(20):9951-61. doi: 10.1128/JVI.01300-06. J Virol. 2006. PMID: 17005673 Free PMC article.
-
Control the host cell cycle: viral regulation of the anaphase-promoting complex.J Virol. 2013 Aug;87(16):8818-25. doi: 10.1128/JVI.00088-13. Epub 2013 Jun 12. J Virol. 2013. PMID: 23760246 Free PMC article. Review.
Cited by
-
Understanding the Cytomegalovirus Cyclin-Dependent Kinase Ortholog pUL97 as a Multifaceted Regulator and an Antiviral Drug Target.Cells. 2024 Aug 13;13(16):1338. doi: 10.3390/cells13161338. Cells. 2024. PMID: 39195228 Free PMC article. Review.
-
Intercellular communication within the virus microenvironment affects the susceptibility of cells to secondary viral infections.Sci Adv. 2023 May 10;9(19):eadg3433. doi: 10.1126/sciadv.adg3433. Epub 2023 May 10. Sci Adv. 2023. PMID: 37163594 Free PMC article.
-
G1/S Cell Cycle Induction by Epstein-Barr Virus BORF2 Is Mediated by P53 and APOBEC3B.J Virol. 2022 Sep 28;96(18):e0066022. doi: 10.1128/jvi.00660-22. Epub 2022 Sep 7. J Virol. 2022. PMID: 36069545 Free PMC article.
-
Studies on the Contribution of Human Cytomegalovirus UL21a and UL97 to Viral Growth and Inactivation of the Anaphase-Promoting Complex/Cyclosome (APC/C) E3 Ubiquitin Ligase Reveal a Unique Cellular Mechanism for Downmodulation of the APC/C Subunits APC1, APC4, and APC5.J Virol. 2015 Jul;89(13):6928-39. doi: 10.1128/JVI.00403-15. Epub 2015 Apr 22. J Virol. 2015. PMID: 25903336 Free PMC article.
-
Human cytomegalovirus riding the cell cycle.Med Microbiol Immunol. 2015 Jun;204(3):409-19. doi: 10.1007/s00430-015-0396-z. Epub 2015 Mar 17. Med Microbiol Immunol. 2015. PMID: 25776080 Review.
References
-
- Britt W (2008) Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol 325: 417–470. - PubMed
-
- Schreiber A, Harter G, Schubert A, Bunjes D, Mertens T, et al. (2009) Antiviral treatment of cytomegalovirus infection and resistant strains. Expert Opin Pharmacother 10: 191–209. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
