

Biogeography and individuality shape function in the human skin metagenome
- PMID: 25279917
- PMCID: PMC4185404
- DOI: 10.1038/nature13786
Biogeography and individuality shape function in the human skin metagenome
Abstract
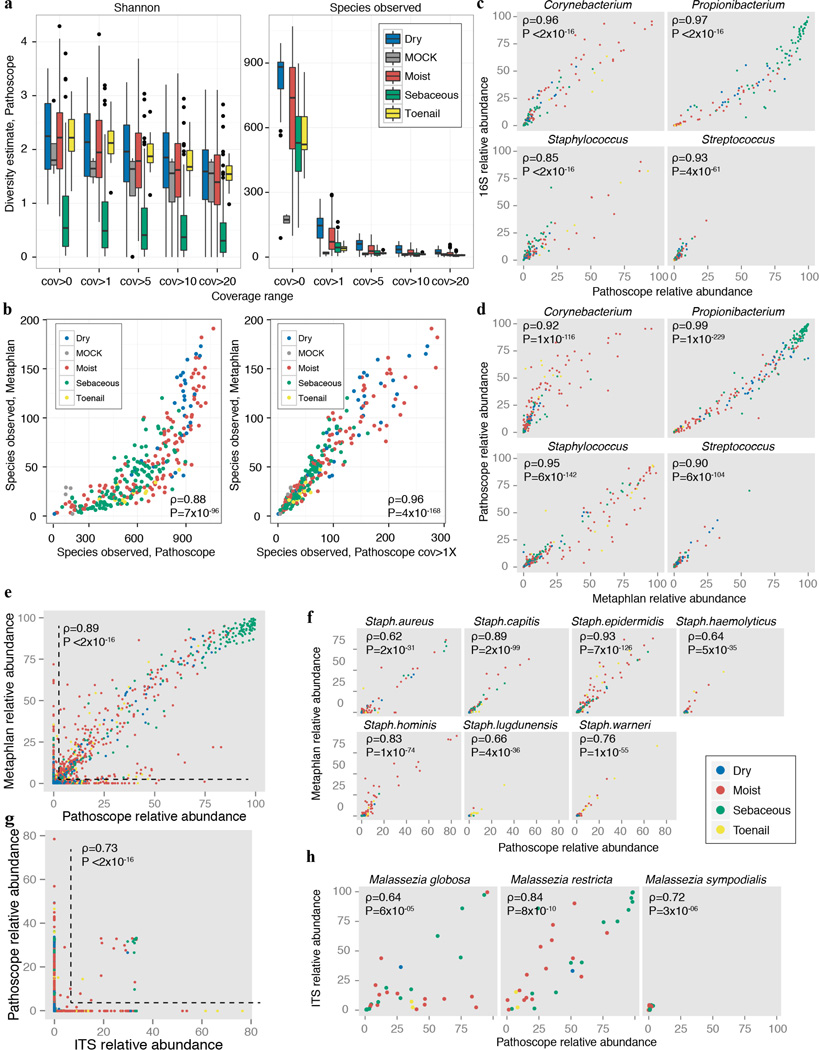
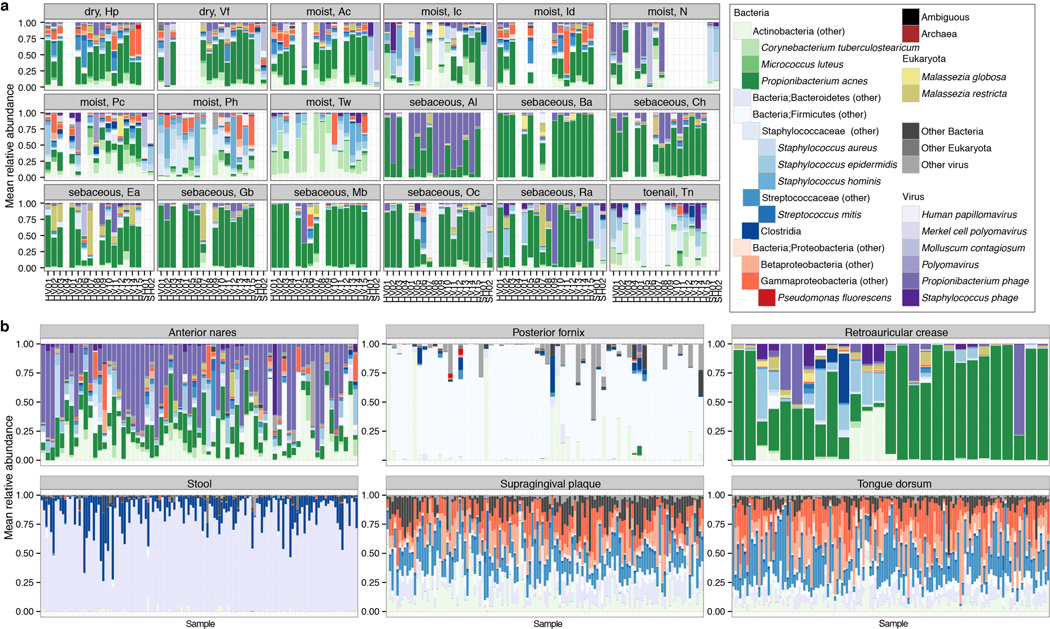
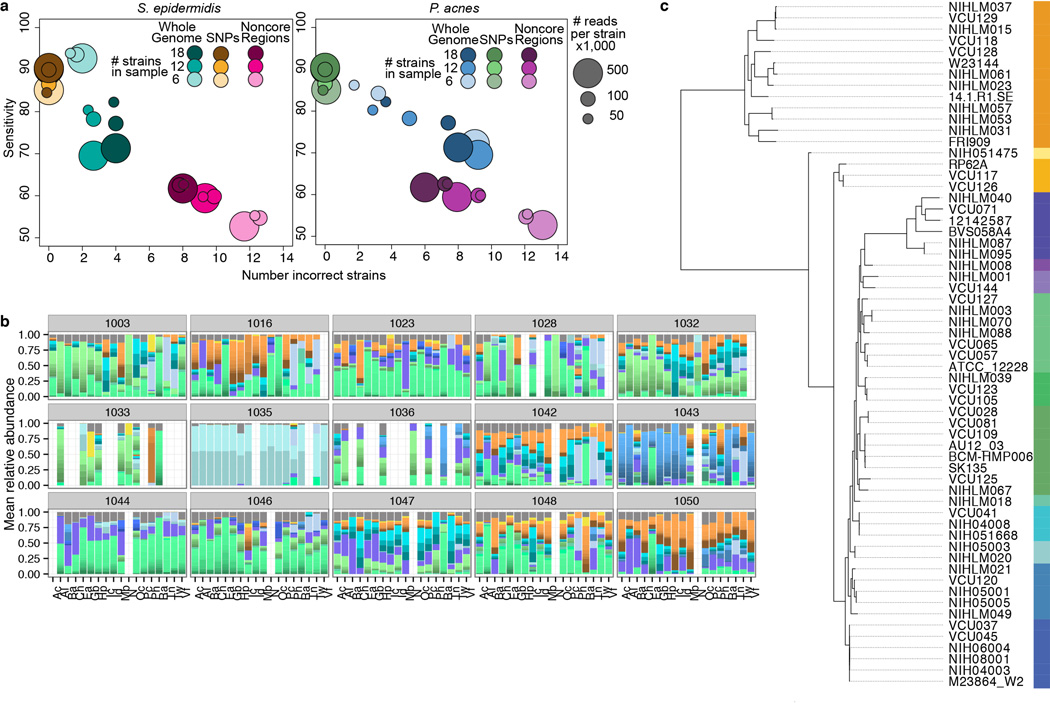
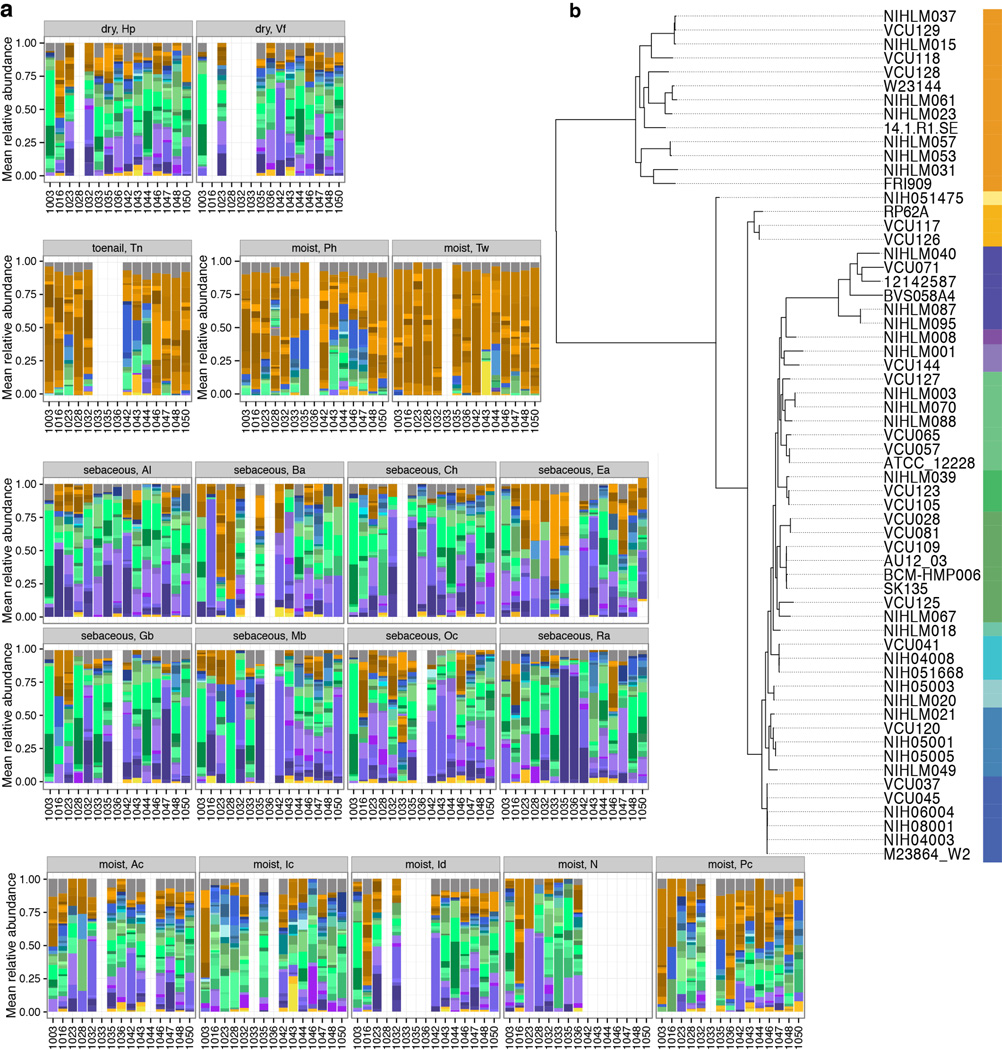
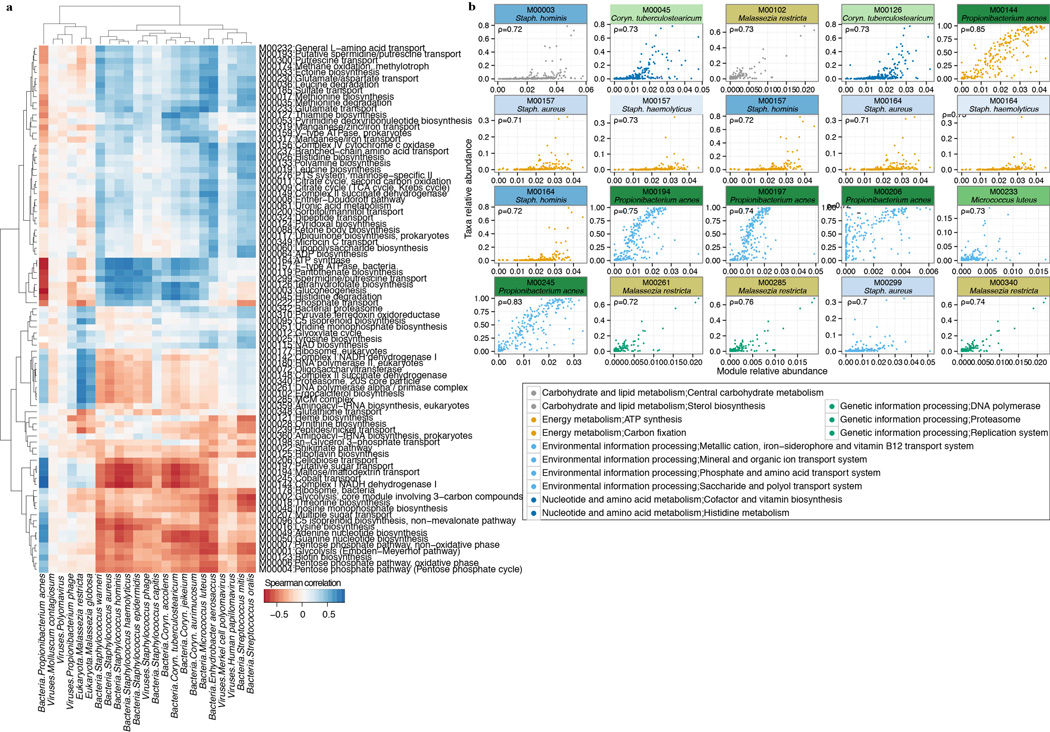
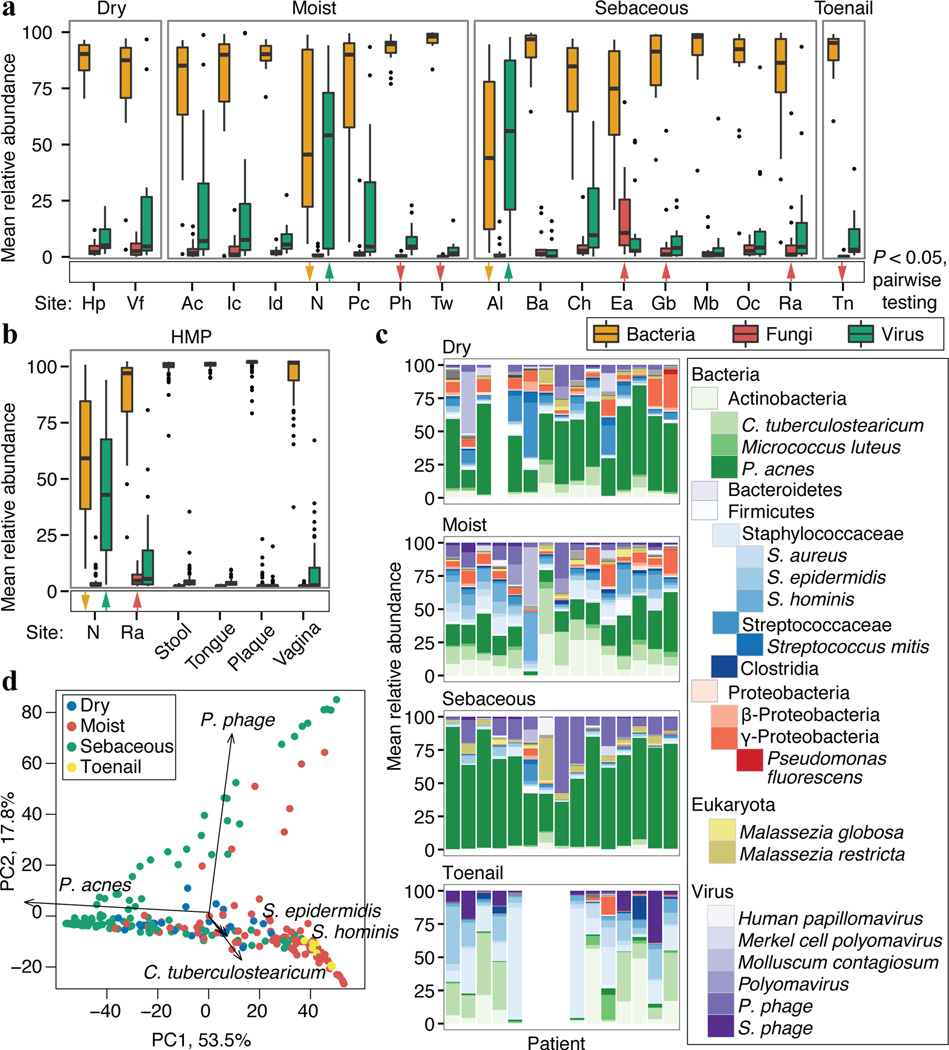
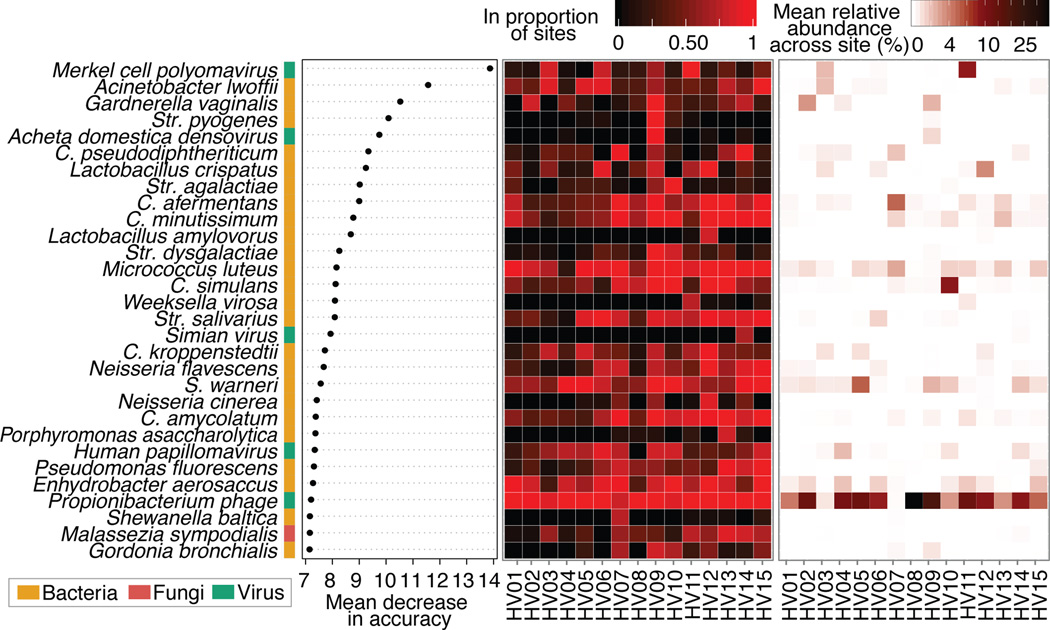
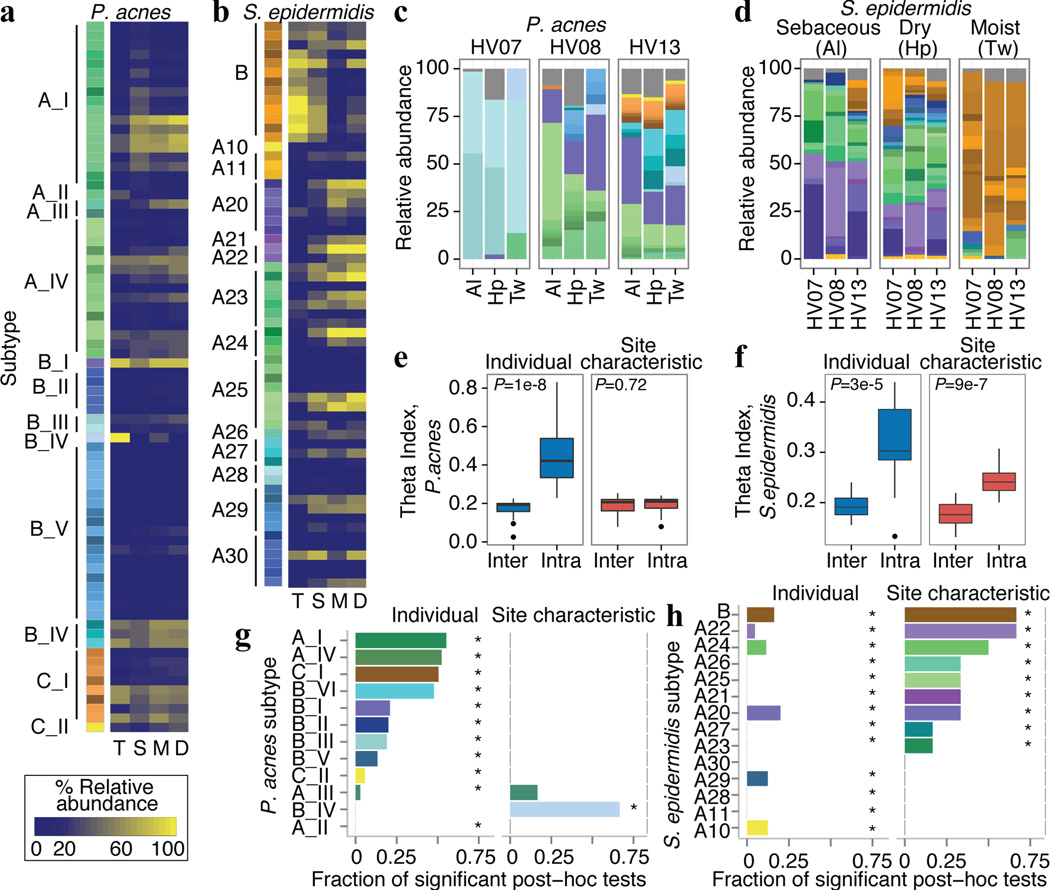
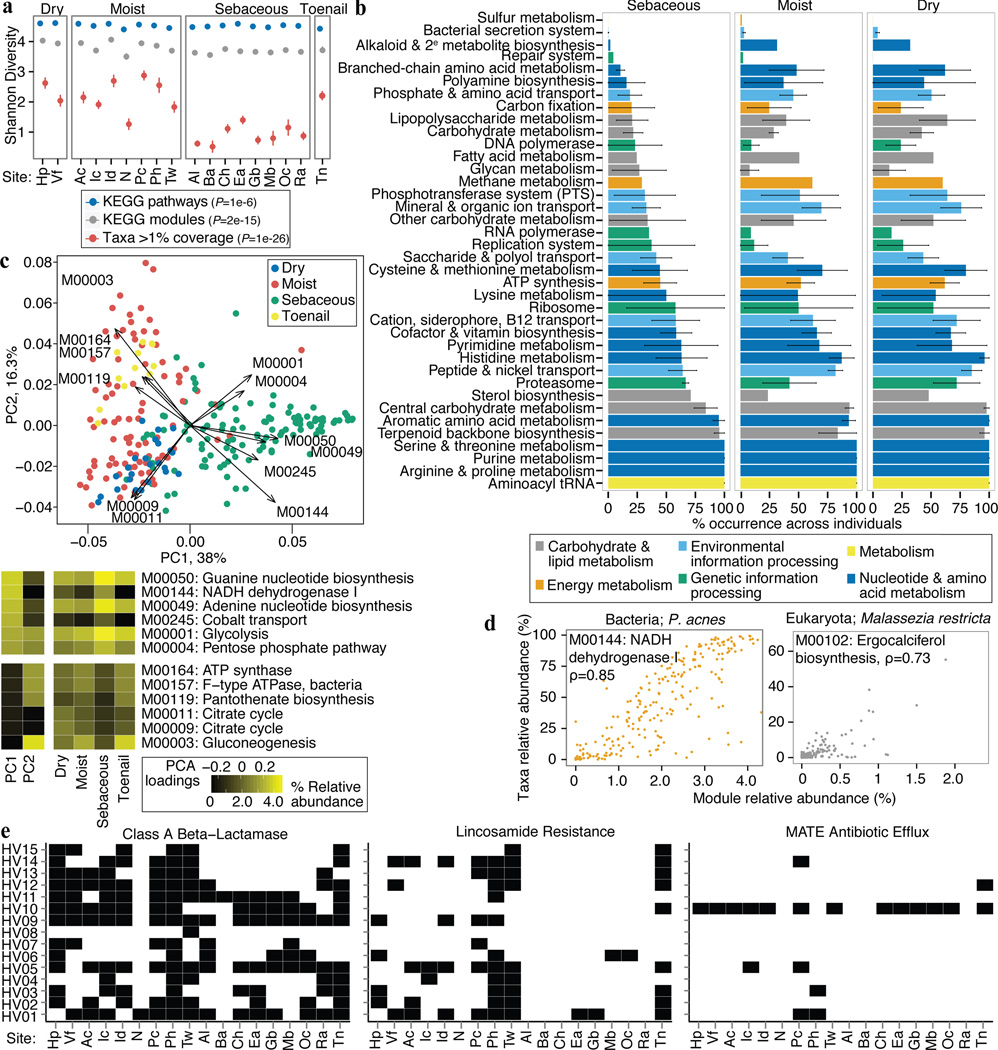
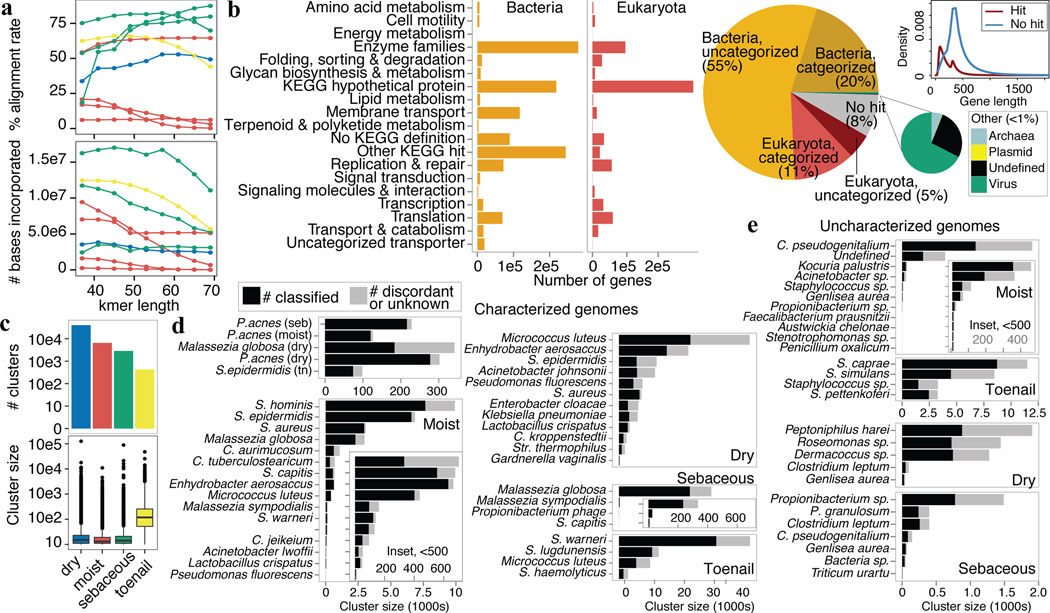
The varied topography of human skin offers a unique opportunity to study how the body's microenvironments influence the functional and taxonomic composition of microbial communities. Phylogenetic marker gene-based studies have identified many bacteria and fungi that colonize distinct skin niches. Here metagenomic analyses of diverse body sites in healthy humans demonstrate that local biogeography and strong individuality define the skin microbiome. We developed a relational analysis of bacterial, fungal and viral communities, which showed not only site specificity but also individual signatures. We further identified strain-level variation of dominant species as heterogeneous and multiphyletic. Reference-free analyses captured the uncharacterized metagenome through the development of a multi-kingdom gene catalogue, which was used to uncover genetic signatures of species lacking reference genomes. This work is foundational for human disease studies investigating inter-kingdom interactions, metabolic changes and strain tracking, and defines the dual influence of biogeography and individuality on microbial composition and function.
Figures





Comment in
-
Microbiology: An integrated view of the skin microbiome.Nature. 2014 Oct 2;514(7520):44-5. doi: 10.1038/514044a. Nature. 2014. PMID: 25279916 No abstract available.
Similar articles
-
Propionibacterium acnes bacteriophages display limited genetic diversity and broad killing activity against bacterial skin isolates.mBio. 2012 Sep 25;3(5):e00279-12. doi: 10.1128/mBio.00279-12. Print 2012. mBio. 2012. PMID: 23015740 Free PMC article.
-
Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization.Genome Res. 2013 Jan;23(1):111-20. doi: 10.1101/gr.142315.112. Epub 2012 Aug 30. Genome Res. 2013. PMID: 22936250 Free PMC article.
-
The diversity and host interactions of Propionibacterium acnes bacteriophages on human skin.ISME J. 2015 Sep;9(9):2078-93. doi: 10.1038/ismej.2015.47. Epub 2015 Apr 7. ISME J. 2015. PMID: 25848871 Free PMC article.
-
Microbiome and skin biology.Curr Opin Allergy Clin Immunol. 2019 Aug;19(4):328-333. doi: 10.1097/ACI.0000000000000542. Curr Opin Allergy Clin Immunol. 2019. PMID: 31107258 Review.
-
Bacterial skin commensals and their role as host guardians.Benef Microbes. 2014 Jun 1;5(2):201-15. doi: 10.3920/BM2012.0062. Benef Microbes. 2014. PMID: 24322878 Review.
Cited by
-
Application of a metatranscriptomics technology, CSI-Dx, for the detection of pathogens associated with prosthetic joint infections.Sci Rep. 2024 Oct 23;14(1):25100. doi: 10.1038/s41598-024-74375-8. Sci Rep. 2024. PMID: 39443495 Free PMC article.
-
The Potential Relevance of the Microbiome to Hair Physiology and Regeneration: The Emerging Role of Metagenomics.Biomedicines. 2021 Feb 26;9(3):236. doi: 10.3390/biomedicines9030236. Biomedicines. 2021. PMID: 33652789 Free PMC article. Review.
-
Strain-Level Diversity Impacts Cheese Rind Microbiome Assembly and Function.mSystems. 2020 Jun 16;5(3):e00149-20. doi: 10.1128/mSystems.00149-20. mSystems. 2020. PMID: 32546667 Free PMC article.
-
A systematic framework for understanding the microbiome in human health and disease: from basic principles to clinical translation.Signal Transduct Target Ther. 2024 Sep 23;9(1):237. doi: 10.1038/s41392-024-01946-6. Signal Transduct Target Ther. 2024. PMID: 39307902 Free PMC article. Review.
-
Chicken skin virome analyzed by high-throughput sequencing shows a composition highly different from human skin.Virus Genes. 2015 Oct;51(2):209-16. doi: 10.1007/s11262-015-1231-8. Epub 2015 Jul 31. Virus Genes. 2015. PMID: 26223320
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
