

The structural basis for cancer treatment decisions
- PMID: 25277176
- PMCID: PMC4202123
- DOI: 10.18632/oncotarget.2439
The structural basis for cancer treatment decisions
Abstract
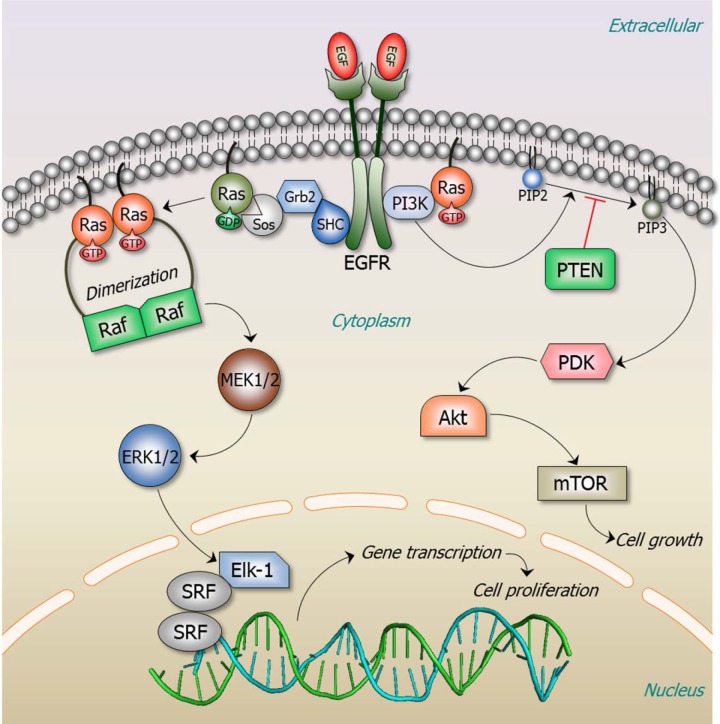
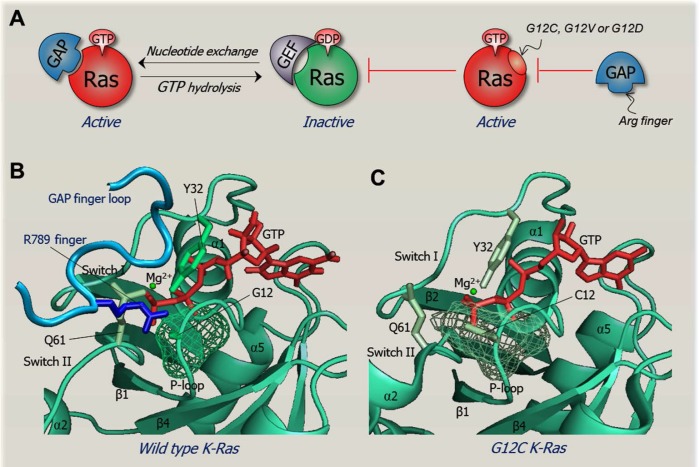
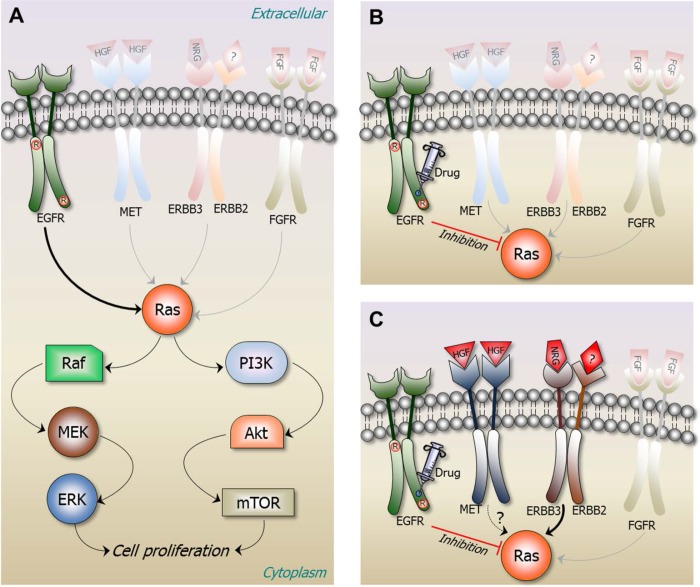
Cancer treatment decisions rely on genetics, large data screens and clinical pharmacology. Here we point out that genetic analysis and treatment decisions may overlook critical elements in cancer development, progression and drug resistance. Two critical structural elements are missing in genetics-based decision-making: the mechanisms of oncogenic mutations and the cellular network which is rewired in cancer. These lay the foundation for the structural basis for cancer treatment decisions, which is rooted in the physical principles of the molecular conformational behavior of single molecules and their interactions. Improved tumor mutational analysis platforms and knowledge of the redundant pathways which can take over in cancer, may not only supplement known actionable findings, but forecast possible cancer progression and resistance. Such forward-looking can be powerful, endowing the oncologist with mechanistic insight and cancer prognosis, and consequently more informed treatment options. Examples include redundant pathways taking over after inhibition of EGFR constitutive activation, mutations in PIK3CA p110α and p85, and the non-hotspot AKT1 mutants conferring constitutive membrane localization.
Figures
Similar articles
-
Hierarchical modeling of activation mechanisms in the ABL and EGFR kinase domains: thermodynamic and mechanistic catalysts of kinase activation by cancer mutations.PLoS Comput Biol. 2009 Aug;5(8):e1000487. doi: 10.1371/journal.pcbi.1000487. Epub 2009 Aug 28. PLoS Comput Biol. 2009. PMID: 19714203 Free PMC article.
-
Human tumor mutants in the p110alpha subunit of PI3K.Cell Cycle. 2006 Apr;5(7):675-7. doi: 10.4161/cc.5.7.2605. Epub 2006 Apr 1. Cell Cycle. 2006. PMID: 16627990 Review.
-
Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Cochrane Database Syst Rev. 2022 Feb 1;2(2022):CD014217. doi: 10.1002/14651858.CD014217. Cochrane Database Syst Rev. 2022. PMID: 36321557 Free PMC article.
-
INTEGRATING GENETIC AND STRUCTURAL DATA ON HUMAN PROTEIN KINOME IN NETWORK-BASED MODELING OF KINASE SENSITIVITIES AND RESISTANCE TO TARGETED AND PERSONALIZED ANTICANCER DRUGS.Pac Symp Biocomput. 2016;21:45-56. Pac Symp Biocomput. 2016. PMID: 26776172
-
Structural effects of oncogenic PI3Kα mutations.Curr Top Microbiol Immunol. 2010;347:43-53. doi: 10.1007/82_2010_53. Curr Top Microbiol Immunol. 2010. PMID: 20593314 Free PMC article. Review.
Cited by
-
Mechanism of activation and the rewired network: New drug design concepts.Med Res Rev. 2022 Mar;42(2):770-799. doi: 10.1002/med.21863. Epub 2021 Oct 25. Med Res Rev. 2022. PMID: 34693559 Free PMC article. Review.
-
A new precision medicine initiative at the dawn of exascale computing.Signal Transduct Target Ther. 2021 Jan 6;6(1):3. doi: 10.1038/s41392-020-00420-3. Signal Transduct Target Ther. 2021. PMID: 33402669 Free PMC article. Review.
-
Mechanistic Insights Into Co-Administration of Allosteric and Orthosteric Drugs to Overcome Drug-Resistance in T315I BCR-ABL1.Front Pharmacol. 2022 Mar 18;13:862504. doi: 10.3389/fphar.2022.862504. eCollection 2022. Front Pharmacol. 2022. PMID: 35370687 Free PMC article.
-
Allostery: Allosteric Cancer Drivers and Innovative Allosteric Drugs.J Mol Biol. 2022 Sep 15;434(17):167569. doi: 10.1016/j.jmb.2022.167569. Epub 2022 Apr 1. J Mol Biol. 2022. PMID: 35378118 Free PMC article. Review.
-
Cell signaling and cancer: a mechanistic insight into drug resistance.Mol Biol Rep. 2019 Oct;46(5):5645-5659. doi: 10.1007/s11033-019-04958-6. Epub 2019 Jul 6. Mol Biol Rep. 2019. PMID: 31280421 Review.
References
-
- Winter C, Kristiansen G, Kersting S, Roy J, Aust D, Knosel T, Rummele P, Jahnke B, Hentrich V, Ruckert F, Niedergethmann M, Weichert W, Bahra M, et al. Google goes cancer: Improving outcome prediction for cancer patients by network-based ranking of marker genes. PLoS Comput Biol. 2012;8:e1002511. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
