

Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels
- PMID: 25233095
- PMCID: PMC4169251
- DOI: 10.1371/journal.pgen.1004663
Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels
Abstract
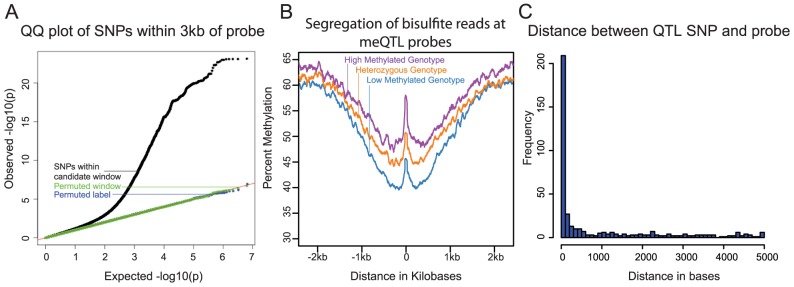
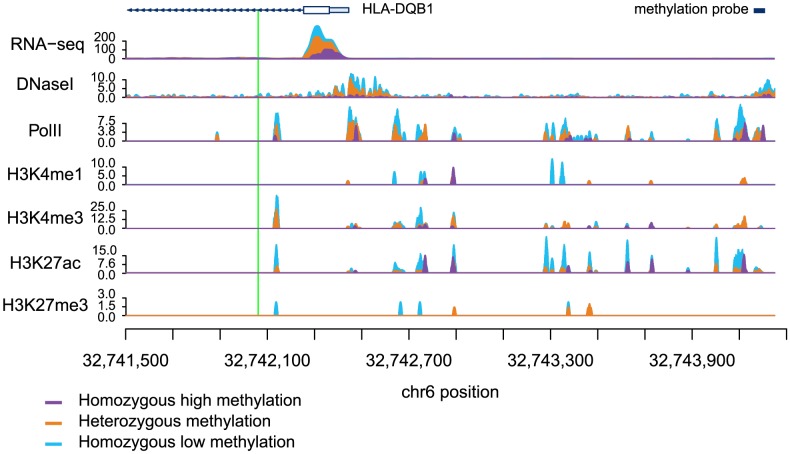
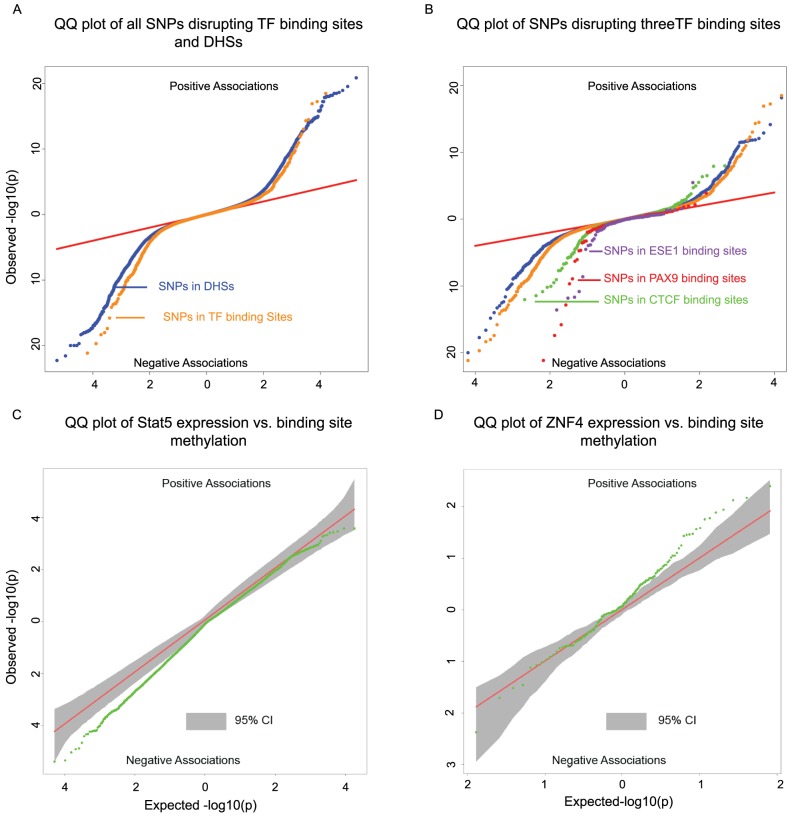
DNA methylation is an important epigenetic regulator of gene expression. Recent studies have revealed widespread associations between genetic variation and methylation levels. However, the mechanistic links between genetic variation and methylation remain unclear. To begin addressing this gap, we collected methylation data at ∼300,000 loci in lymphoblastoid cell lines (LCLs) from 64 HapMap Yoruba individuals, and genome-wide bisulfite sequence data in ten of these individuals. We identified (at an FDR of 10%) 13,915 cis methylation QTLs (meQTLs)-i.e., CpG sites in which changes in DNA methylation are associated with genetic variation at proximal loci. We found that meQTLs are frequently associated with changes in methylation at multiple CpGs across regions of up to 3 kb. Interestingly, meQTLs are also frequently associated with variation in other properties of gene regulation, including histone modifications, DNase I accessibility, chromatin accessibility, and expression levels of nearby genes. These observations suggest that genetic variants may lead to coordinated molecular changes in all of these regulatory phenotypes. One plausible driver of coordinated changes in different regulatory mechanisms is variation in transcription factor (TF) binding. Indeed, we found that SNPs that change predicted TF binding affinities are significantly enriched for associations with DNA methylation at nearby CpGs.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines.Genome Biol. 2011;12(1):R10. doi: 10.1186/gb-2011-12-1-r10. Epub 2011 Jan 20. Genome Biol. 2011. PMID: 21251332 Free PMC article.
-
Characterization of cross-tissue genetic-epigenetic effects and their patterns in schizophrenia.Genome Med. 2018 Feb 26;10(1):13. doi: 10.1186/s13073-018-0519-4. Genome Med. 2018. PMID: 29482655 Free PMC article.
-
Cell-type-specific meQTLs extend melanoma GWAS annotation beyond eQTLs and inform melanocyte gene-regulatory mechanisms.Am J Hum Genet. 2021 Sep 2;108(9):1631-1646. doi: 10.1016/j.ajhg.2021.06.018. Epub 2021 Jul 21. Am J Hum Genet. 2021. PMID: 34293285 Free PMC article.
-
Regulatory SNPs: Altered Transcription Factor Binding Sites Implicated in Complex Traits and Diseases.Int J Mol Sci. 2021 Jun 16;22(12):6454. doi: 10.3390/ijms22126454. Int J Mol Sci. 2021. PMID: 34208629 Free PMC article. Review.
-
Genetic impacts on DNA methylation: research findings and future perspectives.Genome Biol. 2021 Apr 30;22(1):127. doi: 10.1186/s13059-021-02347-6. Genome Biol. 2021. PMID: 33931130 Free PMC article. Review.
Cited by
-
Concordant and discordant DNA methylation signatures of aging in human blood and brain.Epigenetics Chromatin. 2015 May 9;8:19. doi: 10.1186/s13072-015-0011-y. eCollection 2015. Epigenetics Chromatin. 2015. PMID: 25977707 Free PMC article.
-
Allele-specific DNA methylation is increased in cancers and its dense mapping in normal plus neoplastic cells increases the yield of disease-associated regulatory SNPs.Genome Biol. 2020 Jun 29;21(1):153. doi: 10.1186/s13059-020-02059-3. Genome Biol. 2020. PMID: 32594908 Free PMC article.
-
Incorporation of DNA methylation into eQTL mapping in African Americans.Pac Symp Biocomput. 2021;26:244-255. Pac Symp Biocomput. 2021. PMID: 33691021 Free PMC article.
-
Evolution of DNA Methylation in Papio Baboons.Mol Biol Evol. 2019 Mar 1;36(3):527-540. doi: 10.1093/molbev/msy227. Mol Biol Evol. 2019. PMID: 30521003 Free PMC article.
-
Whole Genome DNA Methylation Variations in Mammary Gland Tissues from Holstein Cattle Producing Milk with Various Fat and Protein Contents.Genes (Basel). 2021 Oct 28;12(11):1727. doi: 10.3390/genes12111727. Genes (Basel). 2021. PMID: 34828333 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- UL1 TR000430/TR/NCATS NIH HHS/United States
- GM007197/GM/NIGMS NIH HHS/United States
- HG006123/HG/NHGRI NIH HHS/United States
- R01 MH084703/MH/NIMH NIH HHS/United States
- F31 AG044948/AG/NIA NIH HHS/United States
- R01 HG006123/HG/NHGRI NIH HHS/United States
- F31 AG 044948/AG/NIA NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- DGE-0638477/PHS HHS/United States
- U01 HG007036/HG/NHGRI NIH HHS/United States
- MH084703/MH/NIMH NIH HHS/United States
- T32 GM007197/GM/NIGMS NIH HHS/United States
- U01HG007036/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
