

CEACAM2 negatively regulates hemi (ITAM-bearing) GPVI and CLEC-2 pathways and thrombus growth in vitro and in vivo
- PMID: 25085348
- PMCID: PMC4192753
- DOI: 10.1182/blood-2014-04-569707
CEACAM2 negatively regulates hemi (ITAM-bearing) GPVI and CLEC-2 pathways and thrombus growth in vitro and in vivo
Abstract
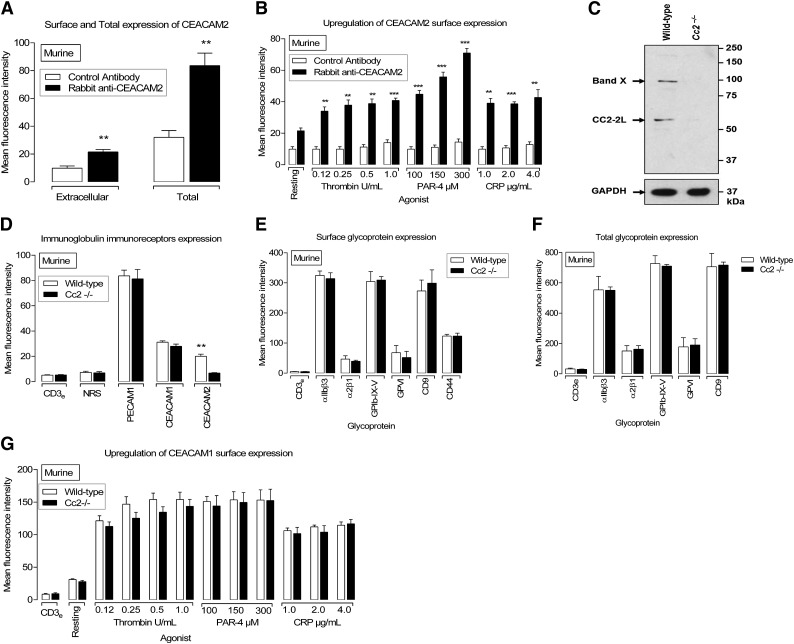
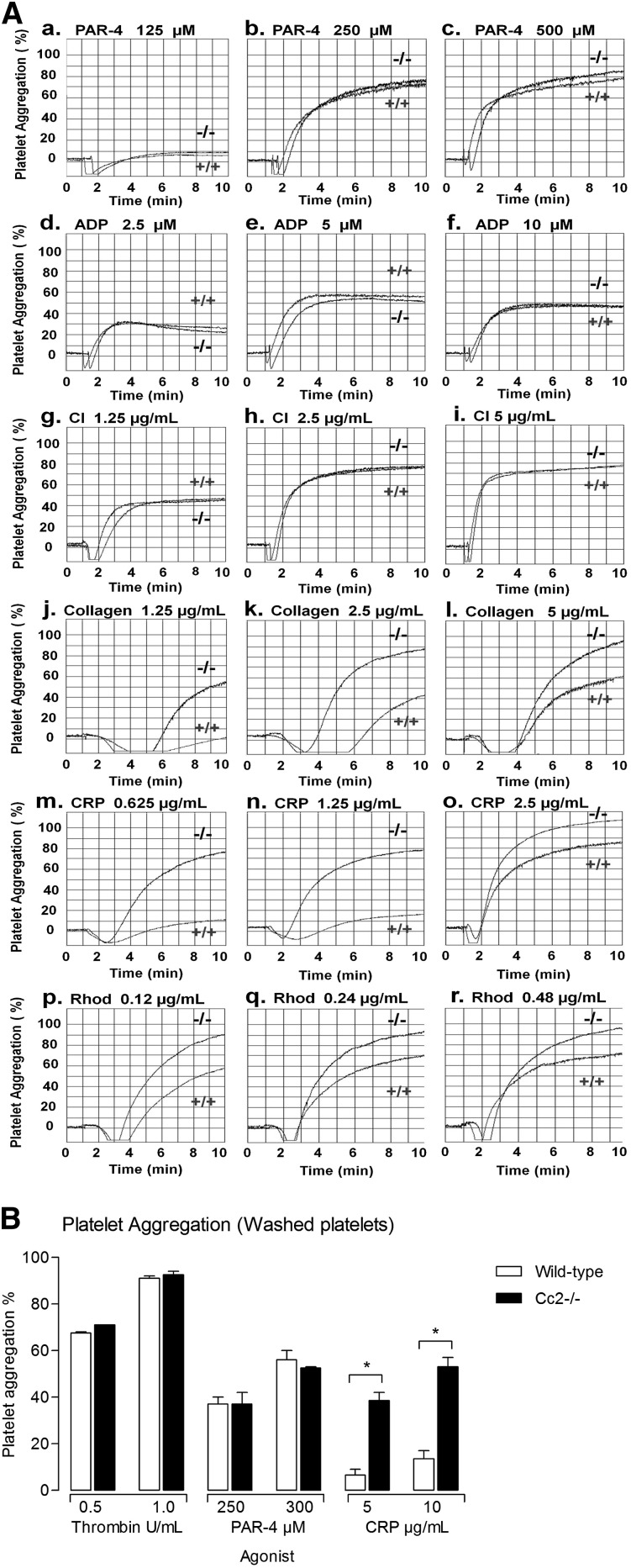
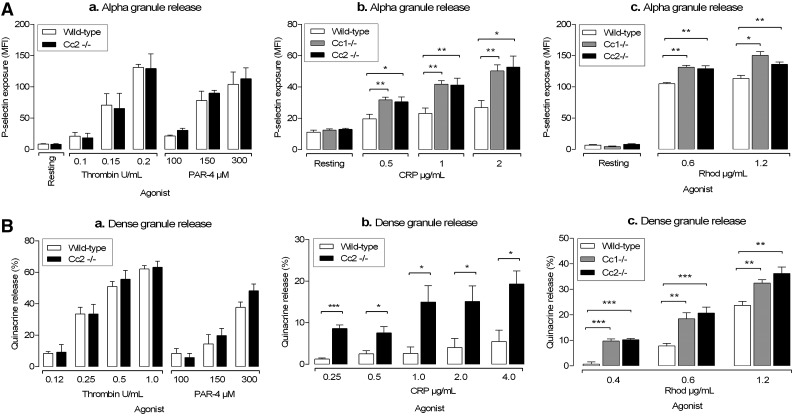
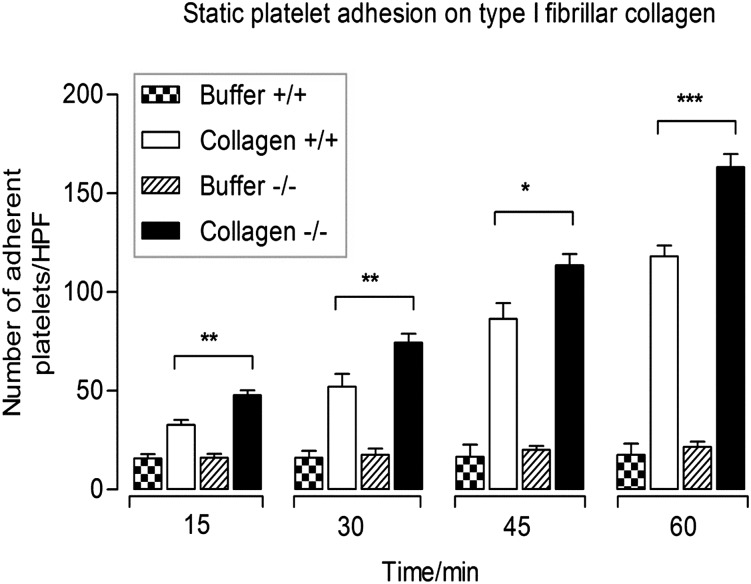
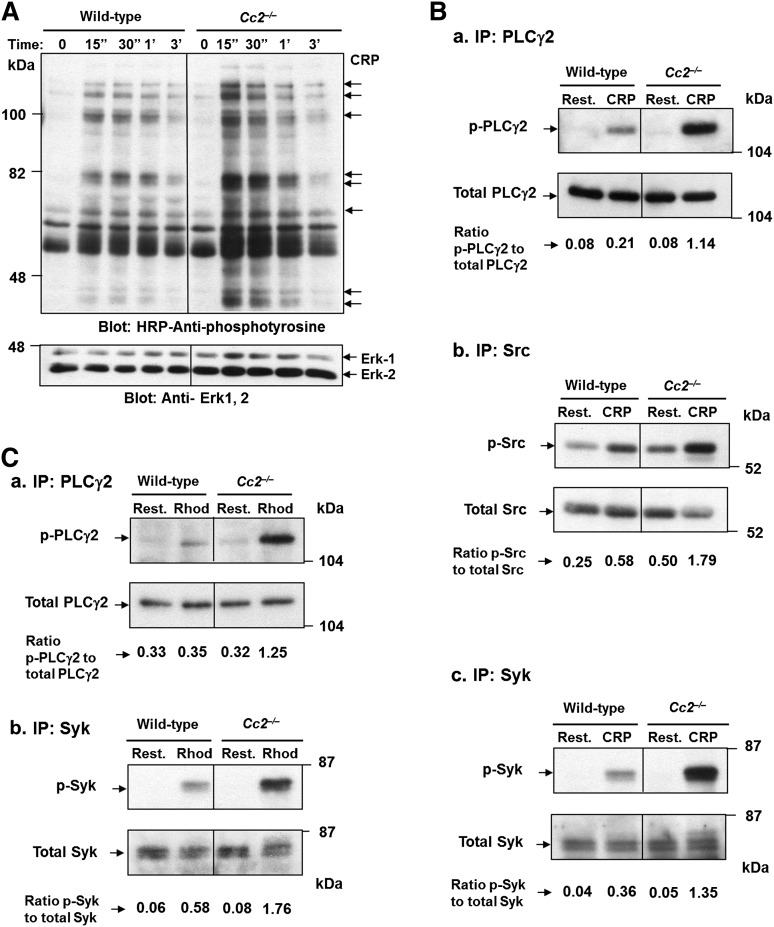
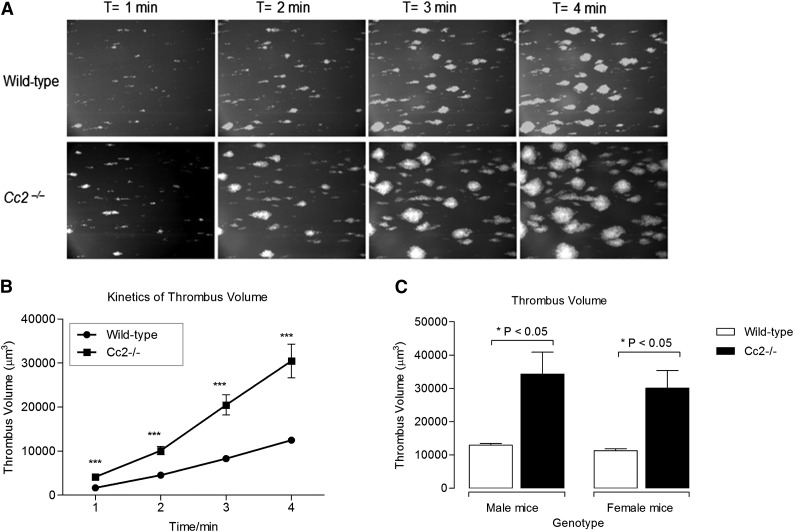
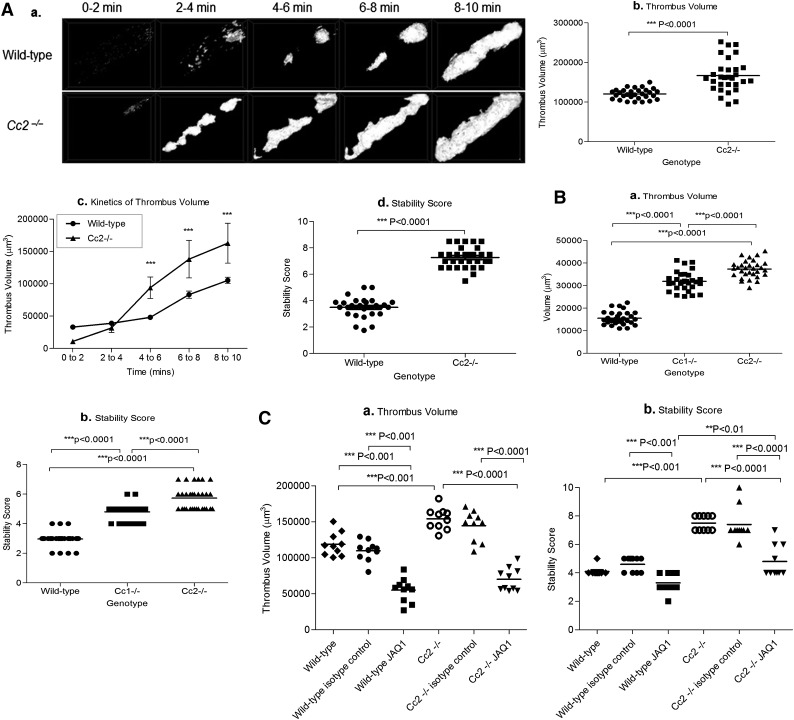
Carcinoembryonic antigen-related cell adhesion molecule-2 (CEACAM2) is a cell-surface glycoprotein expressed on blood, epithelial, and vascular cells. CEACAM2 possesses adhesive and signaling properties mediated by immunoreceptor tyrosine-based inhibitory motifs. In this study, we demonstrate that CEACAM2 is expressed on the surface and in intracellular pools of platelets. Functional studies of platelets from Ceacam2(-/-)-deficient mice (Cc2(-/-)) revealed that CEACAM2 serves to negatively regulate collagen glycoprotein VI (platelet) (GPVI)-FcRγ-chain and the C-type lectinlike receptor 2 (CLEC-2) signaling. Cc2(-/-) platelets displayed enhanced GPVI and CLEC-2-selective ligands, collagen-related peptide (CRP), collagen, and rhodocytin (Rhod)-mediated platelet aggregation. They also exhibited increased adhesion on type I collagen, and hyperresponsive CRP and CLEC-2-induced α and dense granule release compared with wild-type platelets. Furthermore, using intravital microscopy to ferric chloride (FeCl3)-injured mesenteric arterioles and laser-induced injury of cremaster muscle arterioles, we herein show that thrombi formed in Cc2(-/-) mice were larger and more stable than wild-type controls in vivo. Thus, CEACAM2 is a novel platelet immunoreceptor that acts as a negative regulator of platelet GPVI-collagen interactions and of ITAM receptor CLEC-2 pathways.
© 2014 by The American Society of Hematology.
Figures
Comment in
-
CEA a thrombus CAM: CEACAM2, a twin of CEACAM1?Blood. 2014 Oct 9;124(15):2323-4. doi: 10.1182/blood-2014-08-594101. Blood. 2014. PMID: 25301335
Similar articles
-
CEACAM1 negatively regulates platelet-collagen interactions and thrombus growth in vitro and in vivo.Blood. 2009 Feb 19;113(8):1818-28. doi: 10.1182/blood-2008-06-165043. Epub 2008 Nov 13. Blood. 2009. PMID: 19008452
-
CEA a thrombus CAM: CEACAM2, a twin of CEACAM1?Blood. 2014 Oct 9;124(15):2323-4. doi: 10.1182/blood-2014-08-594101. Blood. 2014. PMID: 25301335
-
G6b-B inhibits constitutive and agonist-induced signaling by glycoprotein VI and CLEC-2.J Biol Chem. 2008 Dec 19;283(51):35419-27. doi: 10.1074/jbc.M806895200. Epub 2008 Oct 27. J Biol Chem. 2008. PMID: 18955485 Free PMC article.
-
Novel interactions in platelet biology: CLEC-2/podoplanin and laminin/GPVI.J Thromb Haemost. 2009 Jul;7 Suppl 1:191-4. doi: 10.1111/j.1538-7836.2009.03372.x. J Thromb Haemost. 2009. PMID: 19630798 Review.
-
Platelet immunoreceptor tyrosine-based activation motif (ITAM) and hemITAM signaling and vascular integrity in inflammation and development.J Thromb Haemost. 2016 Apr;14(4):645-54. doi: 10.1111/jth.13250. Epub 2016 Feb 16. J Thromb Haemost. 2016. PMID: 26749528 Review.
Cited by
-
Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction.Platelets. 2016;27(3):230-9. doi: 10.3109/09537104.2015.1083543. Epub 2015 Sep 14. Platelets. 2016. PMID: 26367242 Free PMC article.
-
Age-dependent insulin resistance in male mice with null deletion of the carcinoembryonic antigen-related cell adhesion molecule 2 gene.Diabetologia. 2017 Sep;60(9):1751-1760. doi: 10.1007/s00125-017-4307-2. Epub 2017 May 31. Diabetologia. 2017. PMID: 28567513 Free PMC article.
-
Platelet signaling: a complex interplay between inhibitory and activatory networks.J Thromb Haemost. 2016 May;14(5):918-30. doi: 10.1111/jth.13302. Epub 2016 Apr 9. J Thromb Haemost. 2016. PMID: 26929147 Free PMC article. Review.
-
Carcinoembryonic Cell Adhesion-Related Molecule 2 Regulates Insulin Secretion and Energy Balance.Int J Mol Sci. 2019 Jul 1;20(13):3231. doi: 10.3390/ijms20133231. Int J Mol Sci. 2019. PMID: 31266142 Free PMC article. Review.
-
Negative regulators of platelet activation and adhesion.J Thromb Haemost. 2018 Feb;16(2):220-230. doi: 10.1111/jth.13910. Epub 2017 Dec 26. J Thromb Haemost. 2018. PMID: 29193689 Free PMC article. Review.
References
-
- Jones CI, Barrett NE, Moraes LA, Gibbins JM, Jackson DE. Endogenous inhibitory mechanisms and the regulation of platelet function. Methods Mol Biol. 2012;788:341–366. - PubMed
-
- Jones KL, Hughan SC, Dopheide SM, Farndale RW, Jackson SP, Jackson DE. Platelet endothelial cell adhesion molecule-1 is a negative regulator of platelet-collagen interactions. Blood. 2001;98(5):1456–1463. - PubMed
-
- Patil S, Newman DK, Newman PJ. Platelet endothelial cell adhesion molecule-1 serves as an inhibitory receptor that modulates platelet responses to collagen. Blood. 2001;97(6):1727–1732. - PubMed
-
- Cicmil M, Thomas JM, Leduc M, Bon C, Gibbins JM. Platelet endothelial cell adhesion molecule-1 signaling inhibits the activation of human platelets. Blood. 2002;99(1):137–144. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
