

Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters
- PMID: 25036635
- PMCID: PMC4123684
- DOI: 10.1016/j.cell.2014.06.034
Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters
Abstract
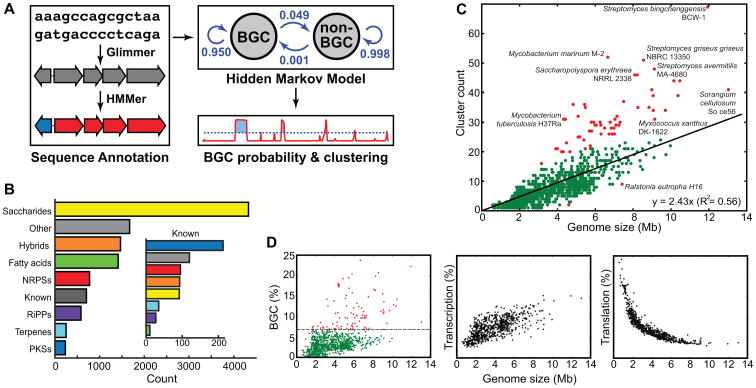
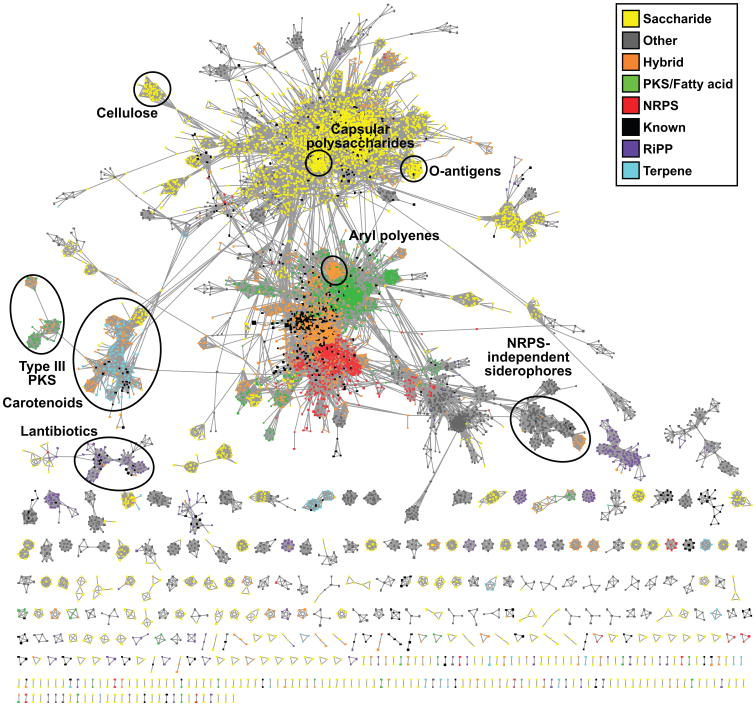
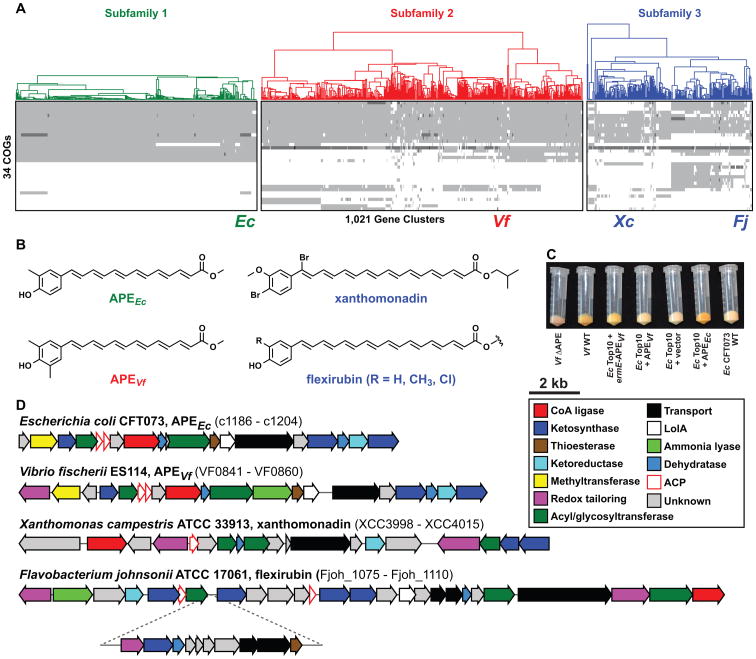
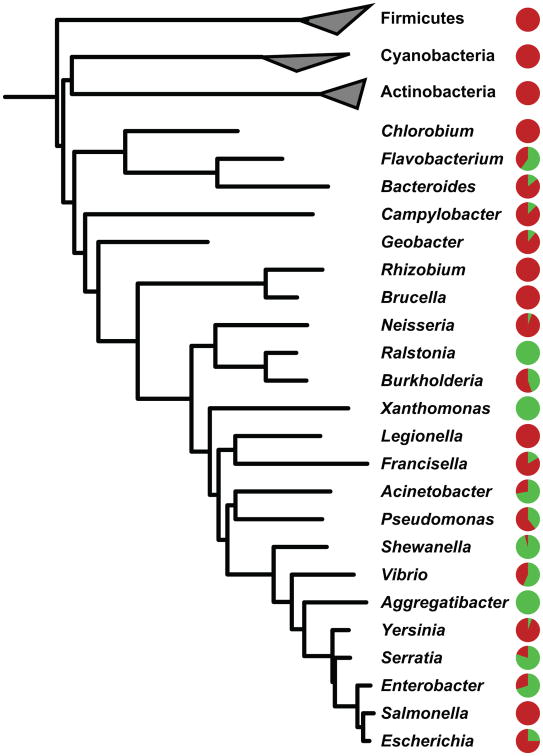
Although biosynthetic gene clusters (BGCs) have been discovered for hundreds of bacterial metabolites, our knowledge of their diversity remains limited. Here, we used a novel algorithm to systematically identify BGCs in the extensive extant microbial sequencing data. Network analysis of the predicted BGCs revealed large gene cluster families, the vast majority uncharacterized. We experimentally characterized the most prominent family, consisting of two subfamilies of hundreds of BGCs distributed throughout the Proteobacteria; their products are aryl polyenes, lipids with an aryl head group conjugated to a polyene tail. We identified a distant relationship to a third subfamily of aryl polyene BGCs, and together the three subfamilies represent the largest known family of biosynthetic gene clusters, with more than 1,000 members. Although these clusters are widely divergent in sequence, their small molecule products are remarkably conserved, indicating for the first time the important roles these compounds play in Gram-negative cell biology.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Biogeography of Bacterial Communities and Specialized Metabolism in Human Aerodigestive Tract Microbiomes.Microbiol Spectr. 2021 Oct 31;9(2):e0166921. doi: 10.1128/Spectrum.01669-21. Epub 2021 Oct 27. Microbiol Spectr. 2021. PMID: 34704787 Free PMC article.
-
Global analyses of biosynthetic gene clusters in phytobiomes reveal strong phylogenetic conservation of terpenes and aryl polyenes.mSystems. 2023 Aug 31;8(4):e0038723. doi: 10.1128/msystems.00387-23. Epub 2023 Jul 6. mSystems. 2023. PMID: 37409823 Free PMC article.
-
Genome Mining Analysis Uncovers the Previously Unknown Biosynthetic Capacity for Secondary Metabolites in Verrucomicrobia.Mar Biotechnol (NY). 2024 Dec;26(6):1324-1335. doi: 10.1007/s10126-024-10374-0. Epub 2024 Sep 24. Mar Biotechnol (NY). 2024. PMID: 39316199
-
Detecting and prioritizing biosynthetic gene clusters for bioactive compounds in bacteria and fungi.Appl Microbiol Biotechnol. 2019 Apr;103(8):3277-3287. doi: 10.1007/s00253-019-09708-z. Epub 2019 Mar 12. Appl Microbiol Biotechnol. 2019. PMID: 30859257 Free PMC article. Review.
-
Activation of silent biosynthetic pathways and discovery of novel secondary metabolites in actinomycetes by co-culture with mycolic acid-containing bacteria.J Ind Microbiol Biotechnol. 2019 Mar;46(3-4):363-374. doi: 10.1007/s10295-018-2100-y. Epub 2018 Nov 28. J Ind Microbiol Biotechnol. 2019. PMID: 30488365 Review.
Cited by
-
Insights into the Biological Properties of Ligands and Identity of Operator Site for LanK Protein Involved in Landomycin Production.Curr Microbiol. 2023 Nov 10;81(1):5. doi: 10.1007/s00284-023-03528-1. Curr Microbiol. 2023. PMID: 37950074
-
Mini review: Genome mining approaches for the identification of secondary metabolite biosynthetic gene clusters in Streptomyces.Comput Struct Biotechnol J. 2020 Jun 21;18:1548-1556. doi: 10.1016/j.csbj.2020.06.024. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 32637051 Free PMC article. Review.
-
antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters.Nucleic Acids Res. 2015 Jul 1;43(W1):W237-43. doi: 10.1093/nar/gkv437. Epub 2015 May 6. Nucleic Acids Res. 2015. PMID: 25948579 Free PMC article.
-
Discovery and overproduction of novel highly bioactive pamamycins through transcriptional engineering of the biosynthetic gene cluster.Microb Cell Fact. 2023 Nov 14;22(1):233. doi: 10.1186/s12934-023-02231-x. Microb Cell Fact. 2023. PMID: 37964282 Free PMC article.
-
Genome analysis provides insights into the biocontrol ability of Mitsuaria sp. strain TWR114.Arch Microbiol. 2021 Aug;203(6):3373-3388. doi: 10.1007/s00203-021-02327-1. Epub 2021 Apr 21. Arch Microbiol. 2021. PMID: 33880605
References
-
- Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. - PMC - PubMed
-
- Barbe V, Vallenet D, Fonknechten N, Kreimeyer A, Oztas S, Labarre L, Cruveiller S, Robert C, Duprat S, Wincker P, et al. Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res. 2004;32:5766–5779. - PMC - PubMed
-
- Bergmann S, Schumann J, Scherlach K, Lange C, Brakhage AA, Hertweck C. Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat Chem Biol. 2007;3:213–217. - PubMed
-
- Challis GL. A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. Chembiochem. 2005;6:601–611. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
