

A virulent strain of deformed wing virus (DWV) of honeybees (Apis mellifera) prevails after Varroa destructor-mediated, or in vitro, transmission
- PMID: 24968198
- PMCID: PMC4072795
- DOI: 10.1371/journal.ppat.1004230
A virulent strain of deformed wing virus (DWV) of honeybees (Apis mellifera) prevails after Varroa destructor-mediated, or in vitro, transmission
Abstract
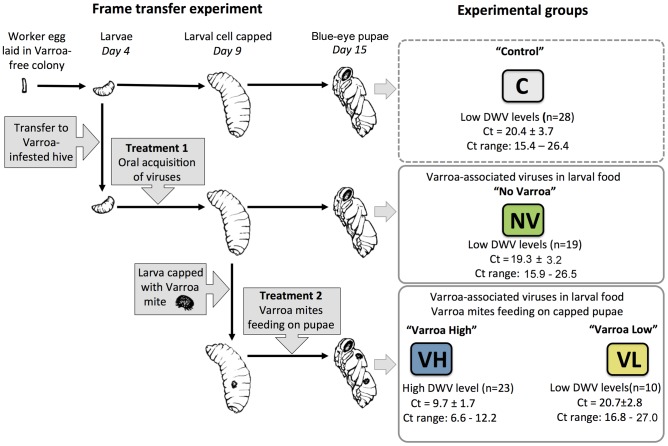
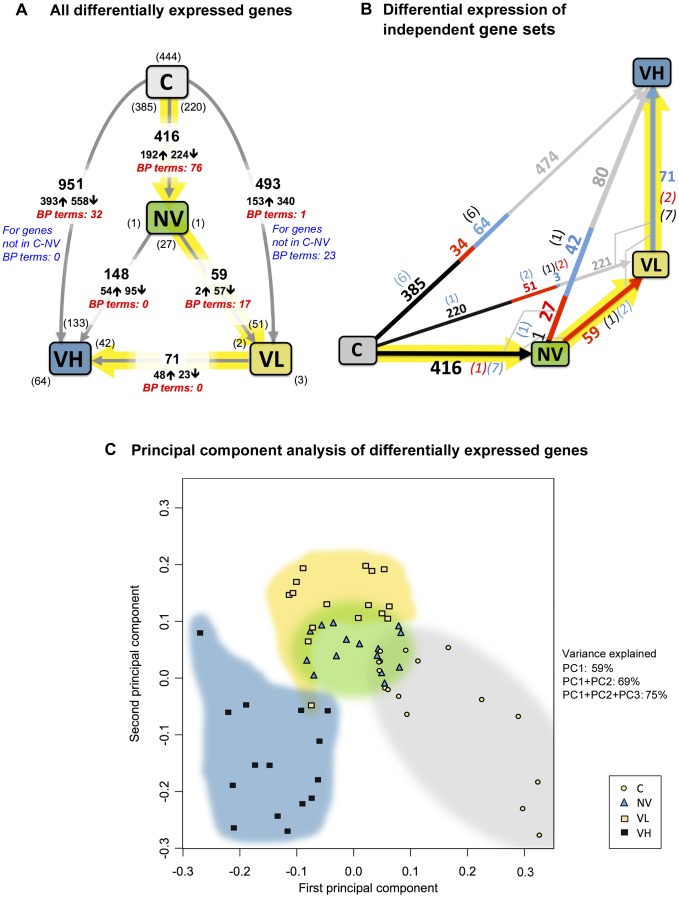
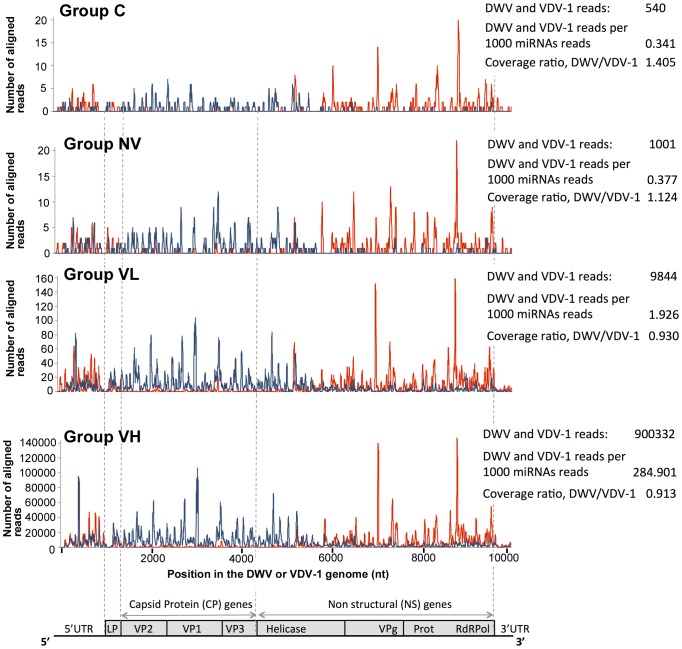
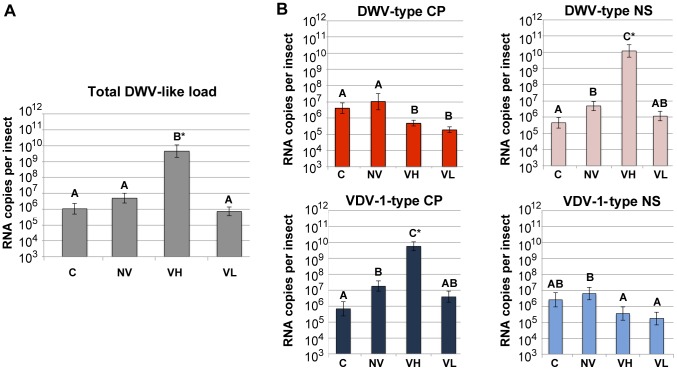
The globally distributed ectoparasite Varroa destructor is a vector for viral pathogens of the Western honeybee (Apis mellifera), in particular the Iflavirus Deformed Wing Virus (DWV). In the absence of Varroa low levels DWV occur, generally causing asymptomatic infections. Conversely, Varroa-infested colonies show markedly elevated virus levels, increased overwintering colony losses, with impairment of pupal development and symptomatic workers. To determine whether changes in the virus population were due Varroa amplifying and introducing virulent virus strains and/or suppressing the host immune responses, we exposed Varroa-naïve larvae to oral and Varroa-transmitted DWV. We monitored virus levels and diversity in developing pupae and associated Varroa, the resulting RNAi response and transcriptome changes in the host. Exposed pupae were stratified by Varroa association (presence/absence) and virus levels (low/high) into three groups. Varroa-free pupae all exhibited low levels of a highly diverse DWV population, with those exposed per os (group NV) exhibiting changes in the population composition. Varroa-associated pupae exhibited either low levels of a diverse DWV population (group VL) or high levels of a near-clonal virulent variant of DWV (group VH). These groups and unexposed controls (C) could be also discriminated by principal component analysis of the transcriptome changes observed, which included several genes involved in development and the immune response. All Varroa tested contained a diverse replicating DWV population implying the virulent variant present in group VH, and predominating in RNA-seq analysis of temporally and geographically separate Varroa-infested colonies, was selected upon transmission from Varroa, a conclusion supported by direct injection of pupae in vitro with mixed virus populations. Identification of a virulent variant of DWV, the role of Varroa in its transmission and the resulting host transcriptome changes furthers our understanding of this important viral pathogen of honeybees.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Direct transmission by injection affects competition among RNA viruses in honeybees.Proc Biol Sci. 2019 Jan 30;286(1895):20182452. doi: 10.1098/rspb.2018.2452. Proc Biol Sci. 2019. PMID: 30963951 Free PMC article.
-
Persistence of subclinical deformed wing virus infections in honeybees following Varroa mite removal and a bee population turnover.PLoS One. 2017 Jul 7;12(7):e0180910. doi: 10.1371/journal.pone.0180910. eCollection 2017. PLoS One. 2017. PMID: 28686725 Free PMC article.
-
Simulated vector transmission differentially influences dynamics of two viral variants of deformed wing virus in honey bees (Apis mellifera).J Gen Virol. 2021 Nov;102(11):001687. doi: 10.1099/jgv.0.001687. J Gen Virol. 2021. PMID: 34816791 Free PMC article.
-
Emerging and re-emerging viruses of the honey bee (Apis mellifera L.).Vet Res. 2010 Nov-Dec;41(6):54. doi: 10.1051/vetres/2010027. Epub 2010 Apr 29. Vet Res. 2010. PMID: 20423694 Free PMC article. Review.
-
The final frontier: ecological and evolutionary dynamics of a global parasite invasion.Biol Lett. 2023 May;19(5):20220589. doi: 10.1098/rsbl.2022.0589. Epub 2023 May 24. Biol Lett. 2023. PMID: 37222245 Free PMC article. Review.
Cited by
-
Changes in the Bacteriome of Honey Bees Associated with the Parasite Varroa destructor, and Pathogens Nosema and Lotmaria passim.Microb Ecol. 2017 Apr;73(3):685-698. doi: 10.1007/s00248-016-0869-7. Epub 2016 Oct 11. Microb Ecol. 2017. PMID: 27730366
-
Overwintering Honey Bee Colonies: Effect of Worker Age and Climate on the Hindgut Microbiota.Insects. 2021 Mar 5;12(3):224. doi: 10.3390/insects12030224. Insects. 2021. PMID: 33807581 Free PMC article.
-
Honey bee viruses in Serbian colonies of different strength.PeerJ. 2018 Nov 14;6:e5887. doi: 10.7717/peerj.5887. eCollection 2018. PeerJ. 2018. PMID: 30479890 Free PMC article.
-
Host density drives viral, but not trypanosome, transmission in a key pollinator.Proc Biol Sci. 2020 Jan 15;287(1918):20191969. doi: 10.1098/rspb.2019.1969. Epub 2020 Jan 8. Proc Biol Sci. 2020. PMID: 31910787 Free PMC article.
-
Contrasting impacts of a novel specialist vector on multihost viral pathogen epidemiology in wild and managed bees.Mol Ecol. 2020 Jan;29(2):380-393. doi: 10.1111/mec.15333. Epub 2020 Jan 7. Mol Ecol. 2020. PMID: 31834965 Free PMC article.
References
-
- Weiss RA (2002) Virulence and pathogenesis. Trends in Microbiology 10: 314–317. - PubMed
-
- Boots M, Greenman J, Ross D, Norman R, Hails R, et al. (2003) The population dynamical implications of covert infections in host-microparasite interactions. Journal of Animal Ecology 72: 1064–1072.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
