

Nonhematopoietic β-Arrestin-1 inhibits inflammation in a murine model of polymicrobial sepsis
- PMID: 24946011
- PMCID: PMC4116700
- DOI: 10.1016/j.ajpath.2014.05.002
Nonhematopoietic β-Arrestin-1 inhibits inflammation in a murine model of polymicrobial sepsis
Abstract
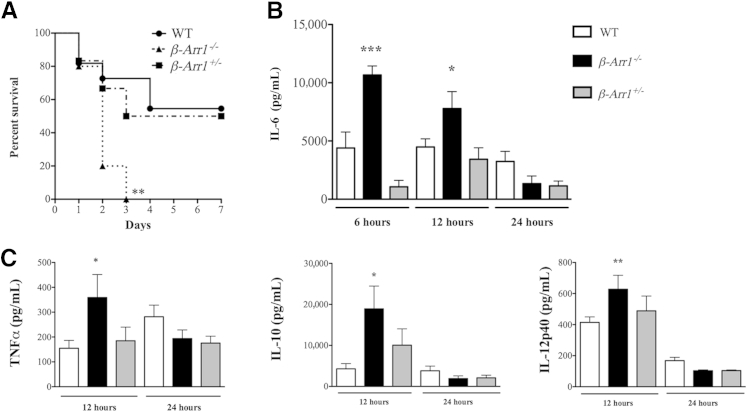
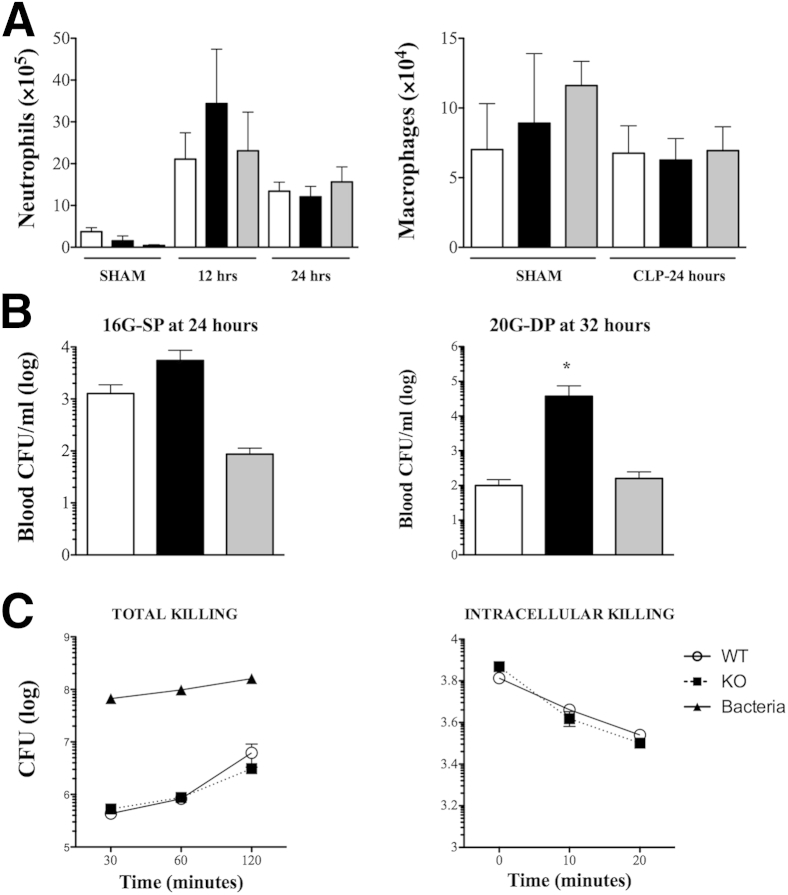
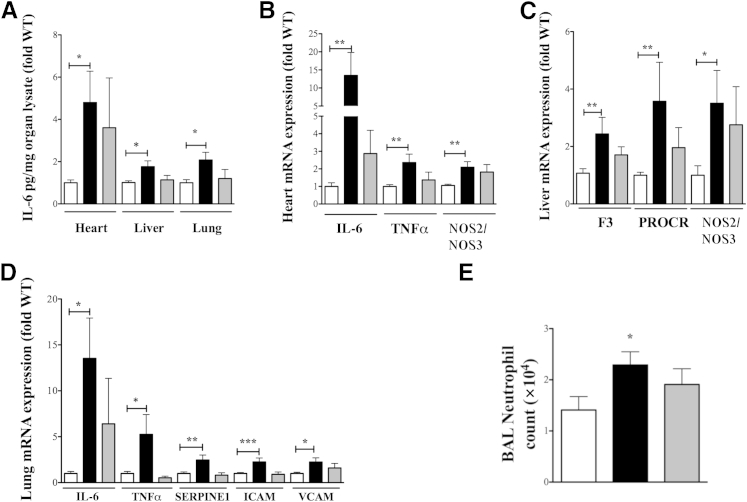
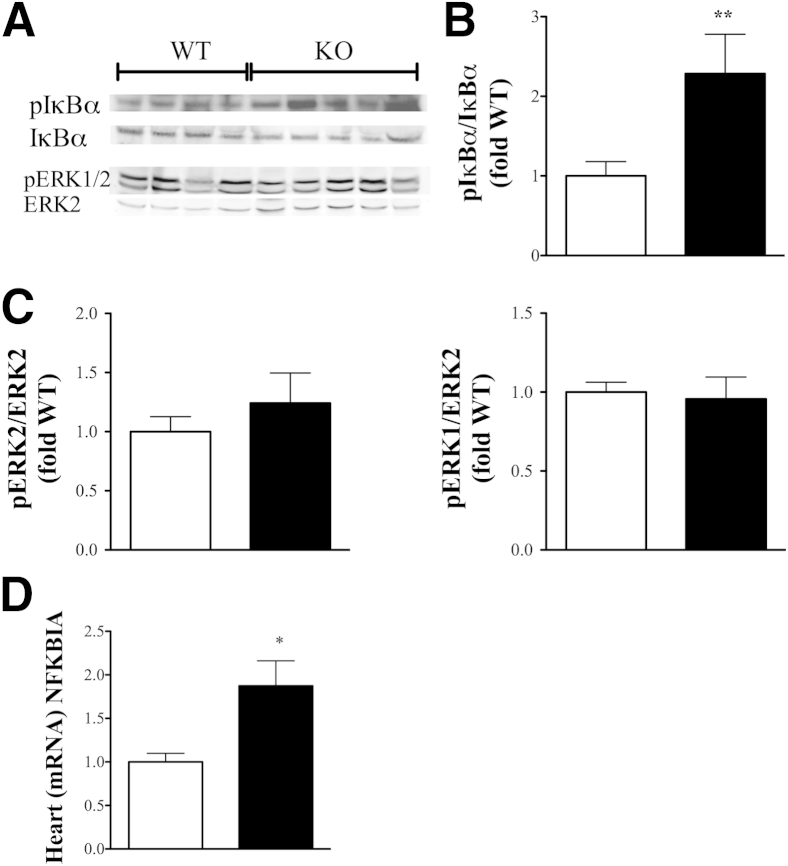
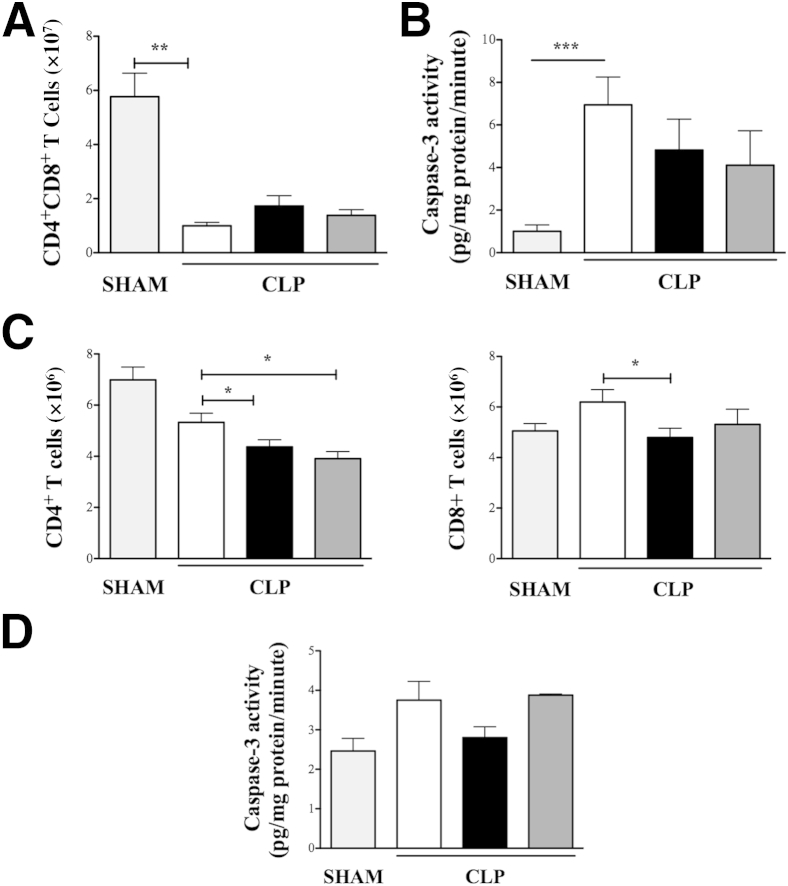
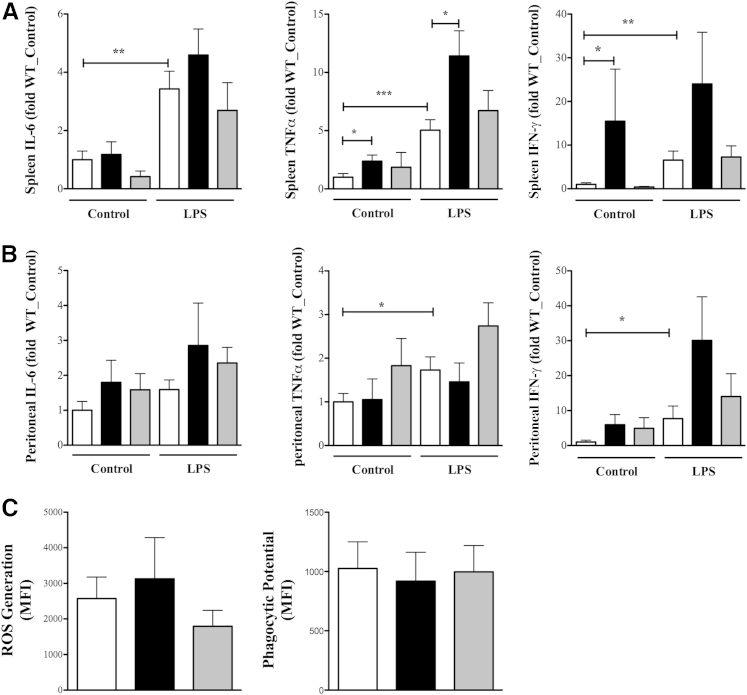
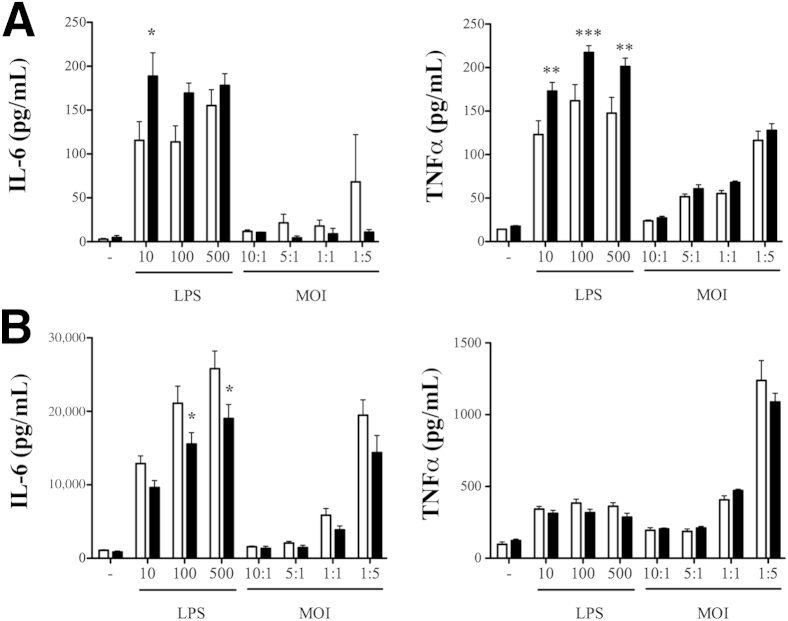
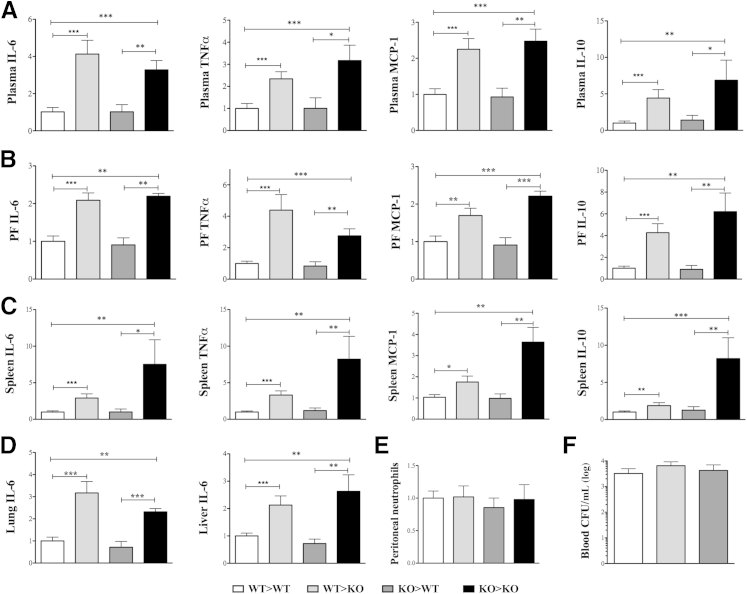
β-Arrestin-1 (βArr1), a scaffolding protein critical in G-protein coupled receptor desensitization has more recently been found to be important in the pathogenesis of various inflammatory diseases. We sought to understand the role of βArr1 in sepsis pathogenesis using a mouse model of polymicrobial sepsis. Although in previous studies we established that βArr1 deficiency protects mice from endotoxemia, here we demonstrate that the absence of βArr1 remarkably renders mice more susceptible to mortality in polymicrobial sepsis. In accordance with the mortality pattern, early production of inflammatory mediators was markedly enhanced in βArr1 knockout mice systemically and locally in various organs. In addition, enhanced inflammation in the heart was associated with increased NFκB activation. Compared to these effects, immune cell infiltration, thymic apoptosis, and immune suppression during polymicrobial sepsis were unaffected by a deficiency of βArr1. Additionally, enhanced inflammation and consequent higher mortality were not observed in heterozygous mice, suggesting that one allele of βArr1 was sufficient for this protective negative regulatory role. We further demonstrate that, unexpectedly, βArr1 in nonhematopoietic cells is critical and sufficient for inhibiting sepsis-induced inflammation, whereas hematopoietic βArr1 is likely redundant. Taken together, our results reveal a novel and previously unrecognized negative regulatory role of the nonhematopoietic βArr1 in sepsis-induced inflammation.
Copyright © 2014 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Gene dosage-dependent negative regulatory role of β-arrestin-2 in polymicrobial infection-induced inflammation.Infect Immun. 2013 Aug;81(8):3035-44. doi: 10.1128/IAI.00653-13. Epub 2013 Jun 10. Infect Immun. 2013. PMID: 23753627 Free PMC article.
-
Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms.Hypertension. 2014 Feb;63(2):404-12. doi: 10.1161/HYPERTENSIONAHA.113.02043. Epub 2013 Nov 11. Hypertension. 2014. PMID: 24218435 Free PMC article.
-
Termination of protease-activated receptor-1 signaling by beta-arrestins is independent of receptor phosphorylation.J Biol Chem. 2004 Mar 12;279(11):10020-31. doi: 10.1074/jbc.M310590200. Epub 2003 Dec 29. J Biol Chem. 2004. PMID: 14699102
-
β-Arrestins 1 and 2 are critical regulators of inflammation.Innate Immun. 2014 Jul;20(5):451-60. doi: 10.1177/1753425913501098. Epub 2013 Sep 12. Innate Immun. 2014. PMID: 24029143 Free PMC article. Review.
-
β-Arrestins and G protein-coupled receptor trafficking.Methods Enzymol. 2013;521:91-108. doi: 10.1016/B978-0-12-391862-8.00005-3. Methods Enzymol. 2013. PMID: 23351735 Review.
Cited by
-
Inflammatory cytokine expression in patients with sepsis at an intensive care unit.Exp Ther Med. 2018 Sep;16(3):2126-2131. doi: 10.3892/etm.2018.6376. Epub 2018 Jun 29. Exp Ther Med. 2018. PMID: 30186449 Free PMC article.
-
ARRB1 downregulates acetaminophen-induced hepatoxicity through binding to p-eIF2α to inhibit ER stress signaling.Cell Biol Toxicol. 2024 Jan 22;40(1):1. doi: 10.1007/s10565-024-09842-z. Cell Biol Toxicol. 2024. PMID: 38252352 Free PMC article.
-
ARRB1 suppresses the activation of hepatic macrophages via modulating endoplasmic reticulum stress in lipopolysaccharide-induced acute liver injury.Cell Death Discov. 2021 Aug 28;7(1):223. doi: 10.1038/s41420-021-00615-9. Cell Death Discov. 2021. PMID: 34455423 Free PMC article.
-
β-Arrestin1 inhibits chemotherapy-induced intestinal stem cell apoptosis and mucositis.Cell Death Dis. 2016 May 19;7(5):e2229. doi: 10.1038/cddis.2016.136. Cell Death Dis. 2016. PMID: 27195676 Free PMC article.
-
Characterization of lncRNA-Based ceRNA Network and Potential Prognostic Hub Genes for Sepsis.Evid Based Complement Alternat Med. 2022 Jun 21;2022:1485033. doi: 10.1155/2022/1485033. eCollection 2022. Evid Based Complement Alternat Med. 2022. Retraction in: Evid Based Complement Alternat Med. 2023 Jun 21;2023:9851697. doi: 10.1155/2023/9851697 PMID: 35774747 Free PMC article. Retracted.
References
-
- Papathanassoglou E.D., Moynihan J.A., Ackerman M.H. Does programmed cell death (apoptosis) play a role in the development of multiple organ dysfunction in critically ill patients? A review and a theoretical framework. Crit Care Med. 2000;28:537–549. - PubMed
-
- Shenoy S.K. Seven-transmembrane receptor signaling through beta-arrestin. Sci STKE. 2005;2005:cm10. - PubMed
-
- DeFea K.A. Stop that cell! [Beta]-arrestin-dependent chemotaxis: a tale of localized actin assembly and receptor desensitization. Annu Rev Physiol. 2007;69:535–560. - PubMed
-
- Luttrell L., Ferguson S., Daaka Y., Miller W., Maudsley S., Della Rocca G., Lin F.T., Kawakatsu H., Owada K., Luttrell D. [Beta]-Arrestin-dependent formation of β2-adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. - PubMed
-
- Violin J.D., DeWire S.M., Yamashita D., Rominger D.H., Nguyen L., Schiller K., Whalen E.J., Gowen M., Lark M.W. Selectively engaging [beta]-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther. 2010;335:572–579. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
