

Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib
- PMID: 24939851
- PMCID: PMC4118115
- DOI: 10.1074/jbc.M114.558890
Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib
Abstract
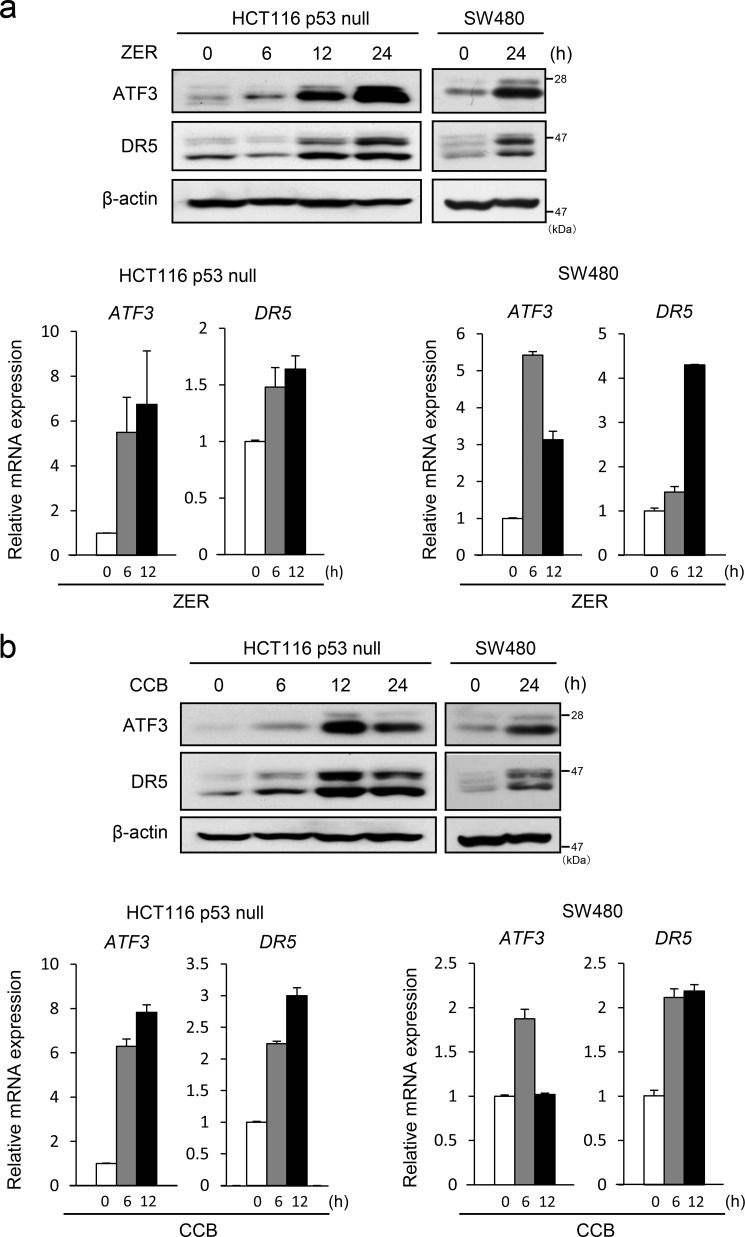
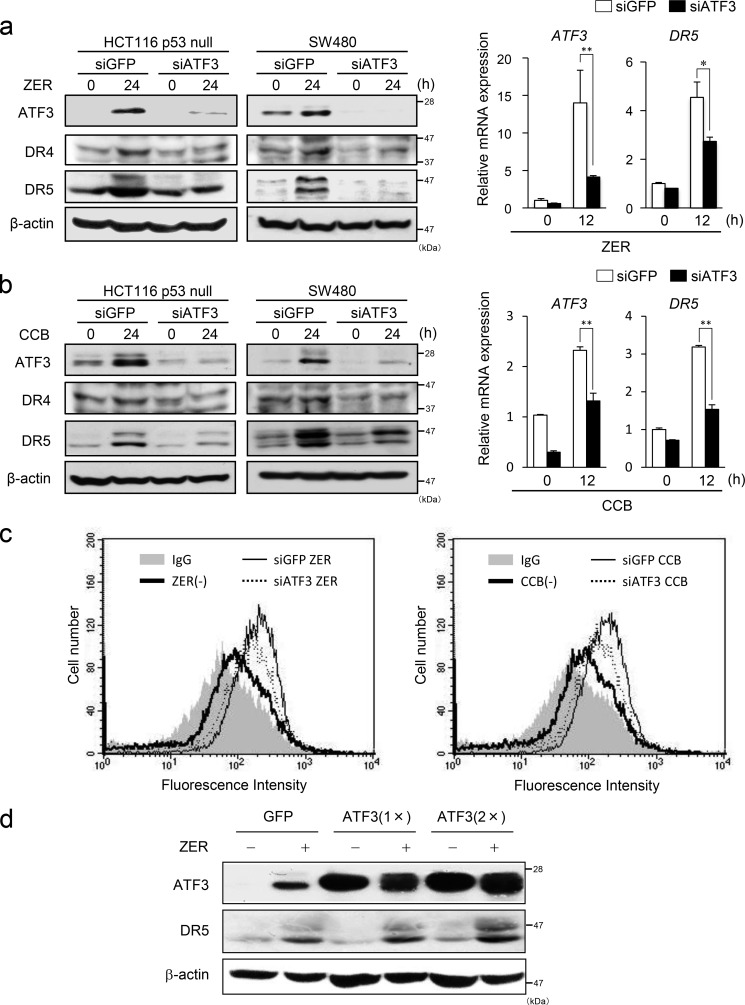
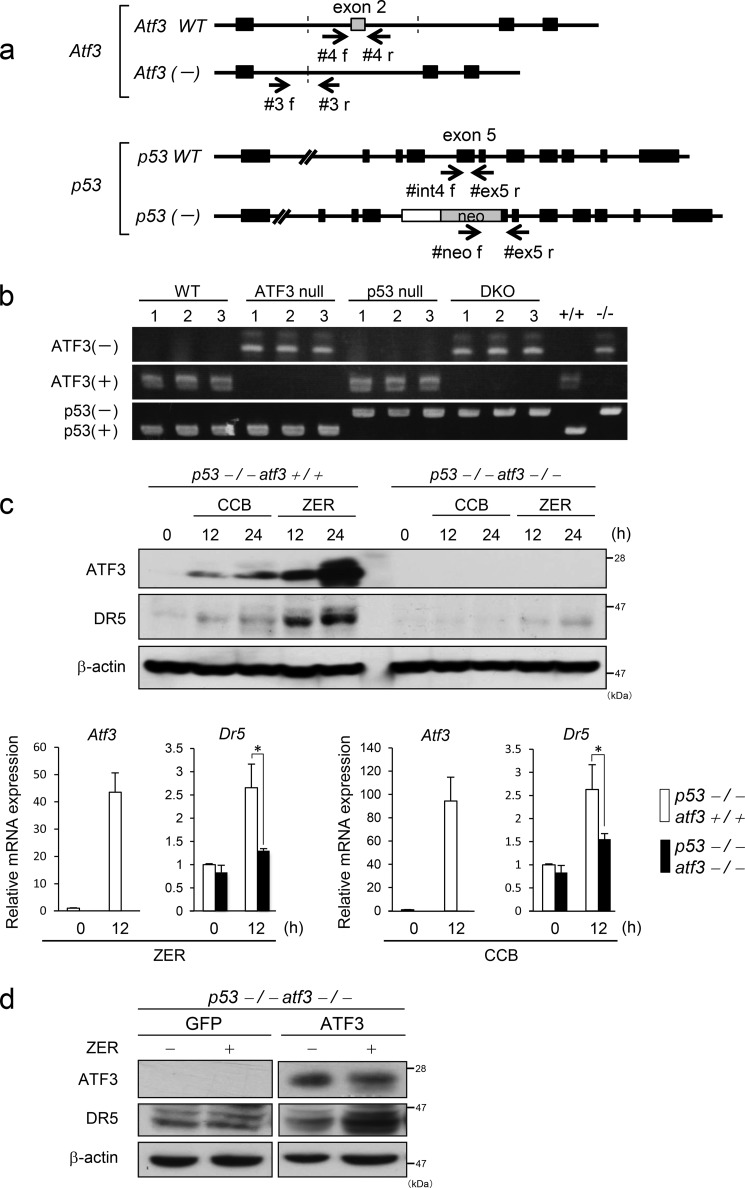
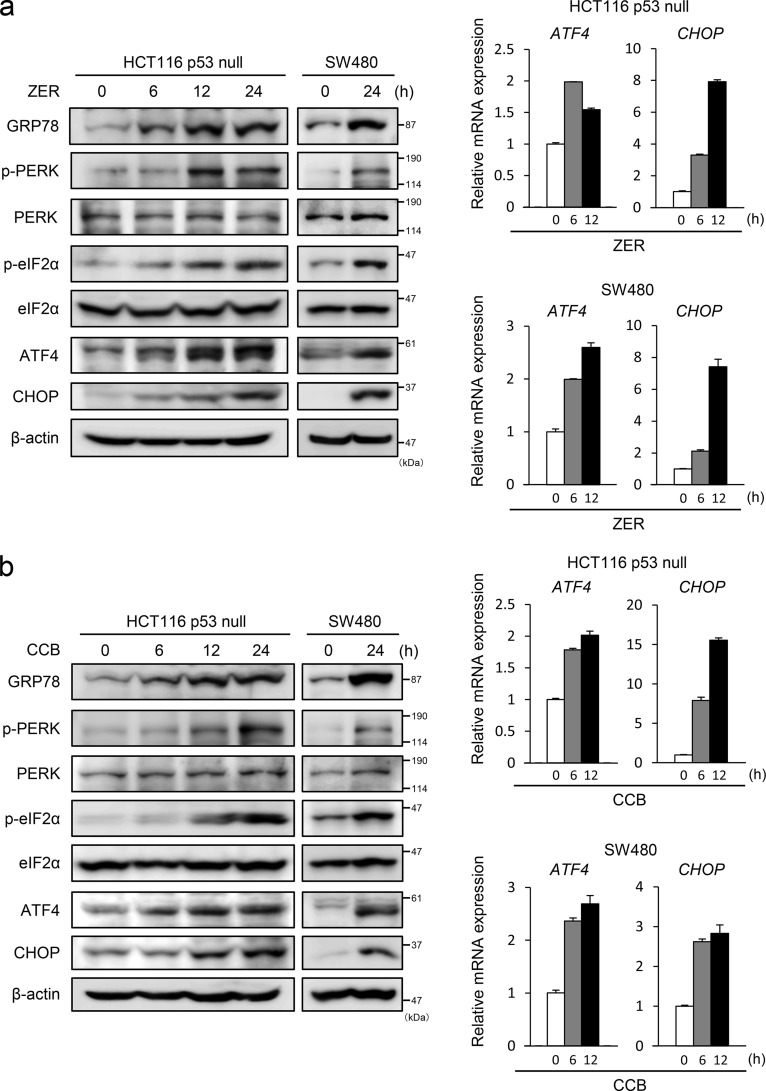
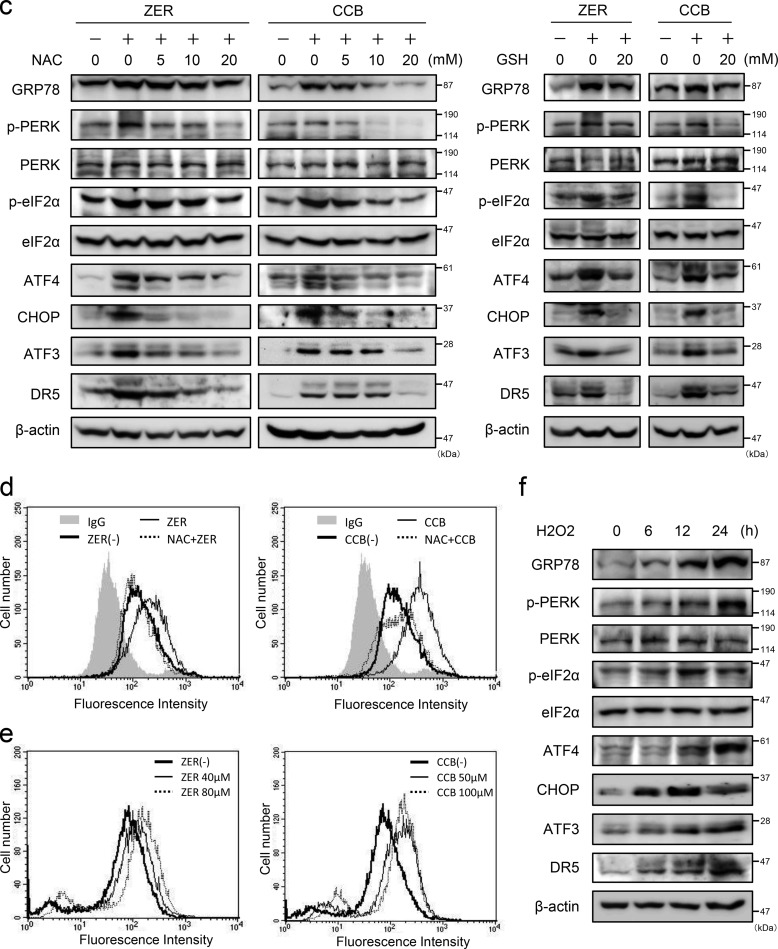
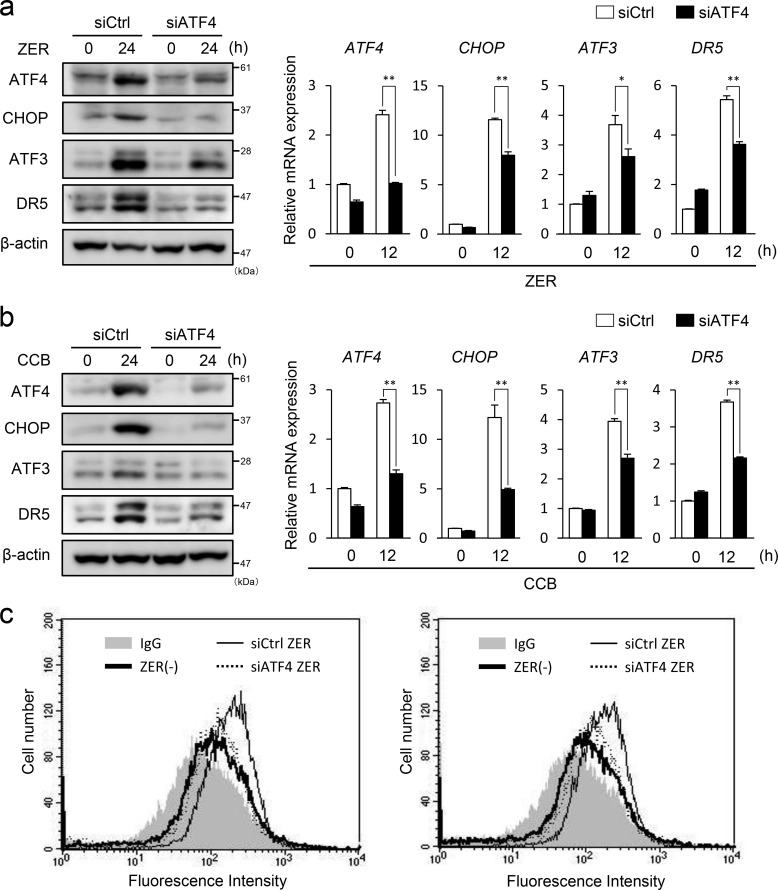
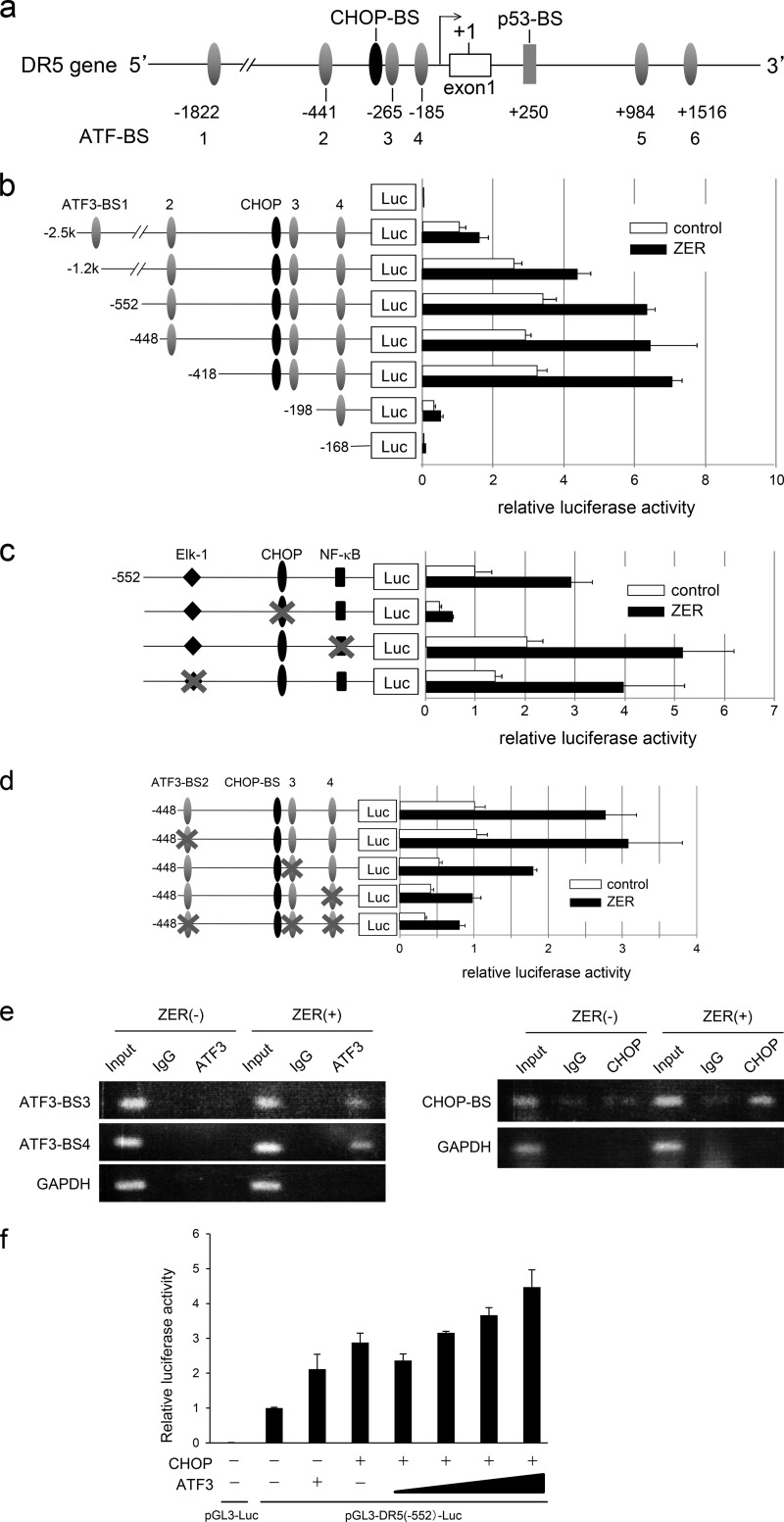
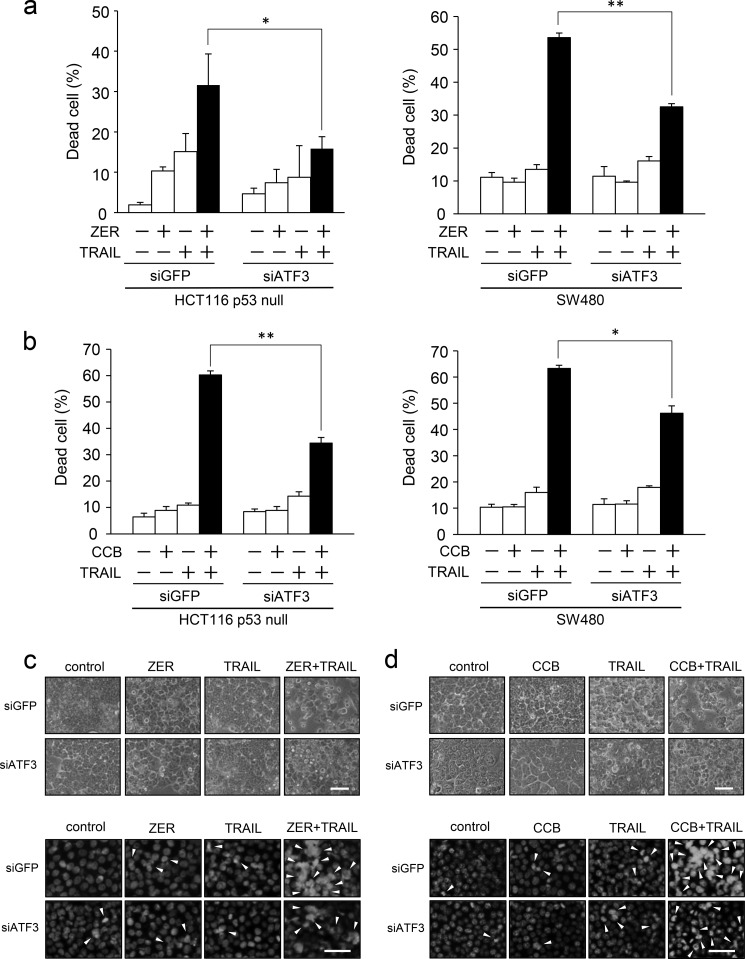
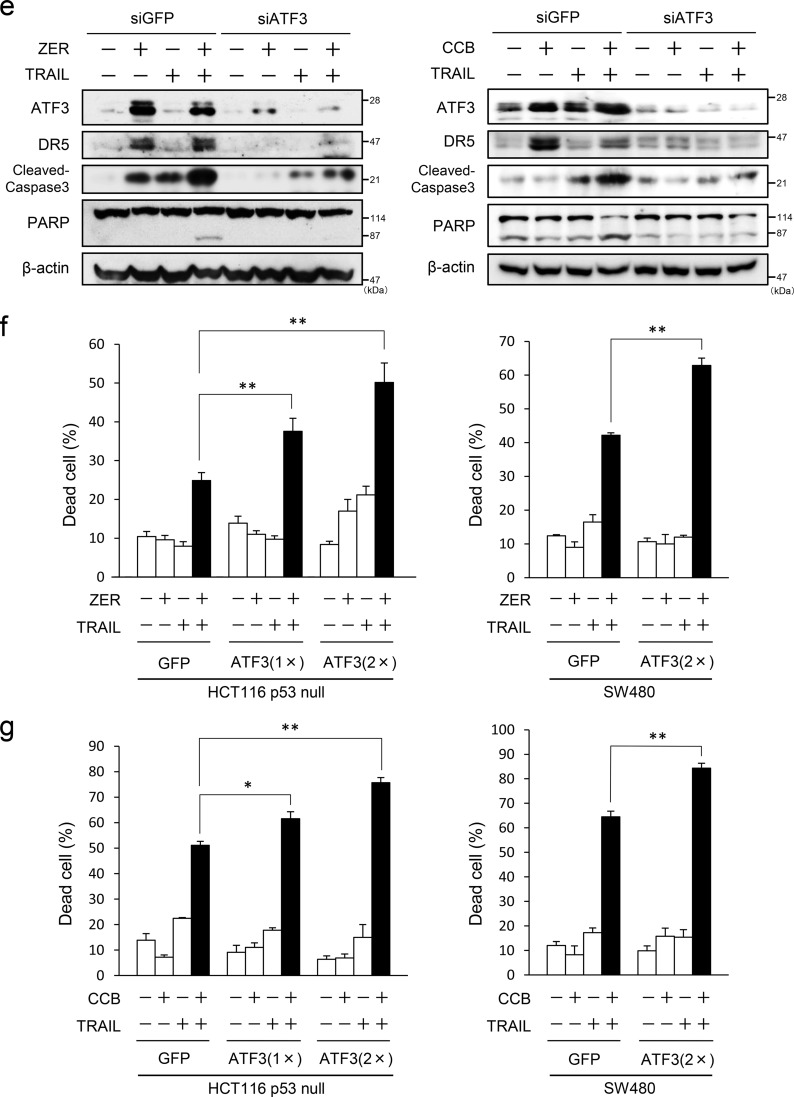
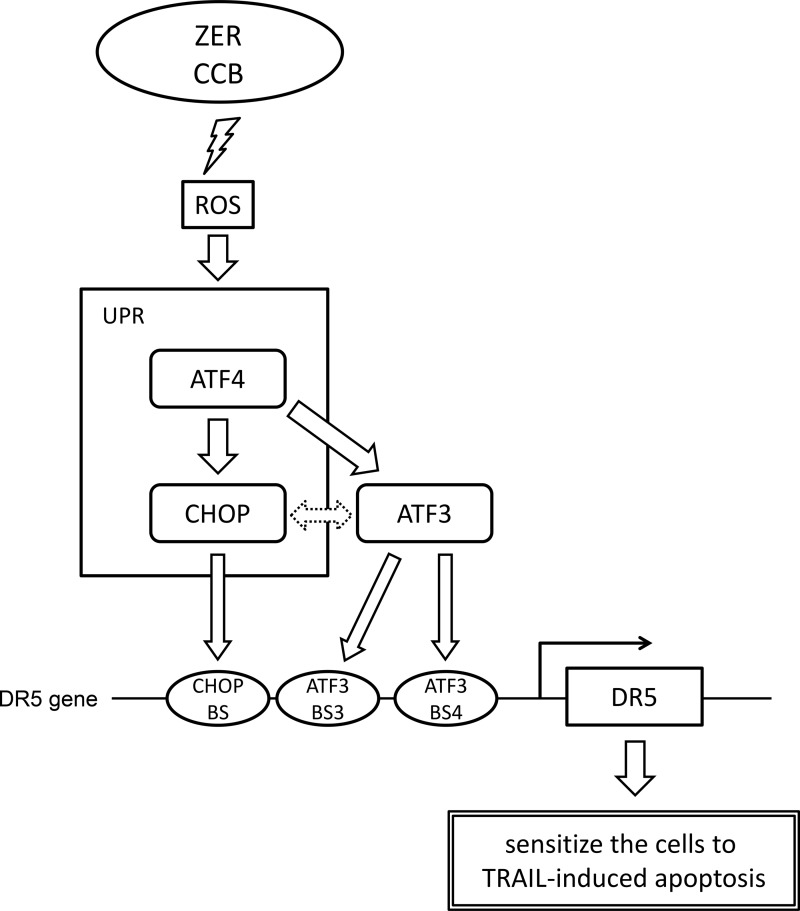
Death receptor 5 (DR5) is a death domain-containing transmembrane receptor that triggers cell death upon binding to its ligand, TNF-related apoptosis-inducing ligand (TRAIL), and a combination of TRAIL and agents that increase the expression of DR5 is expected to be a novel anticancer therapy. In this report, we demonstrate that the stress response gene ATF3 is required for endoplasmic reticulum stress-mediated DR5 induction upon zerumbone (ZER) and celecoxib (CCB) in human p53-deficient colorectal cancer cells. Both agents activated PERK-eIF2α kinases and induced the expression of activating transcription factor 4 (ATF4)-CCAAT enhancer-binding protein (C/EBP) homologous protein, which were remarkably suppressed by reactive oxygen species scavengers. In the absence of ATF3, the induction of DR5 mRNA and protein was abrogated significantly, and this was associated with reduced cell death by cotreatment of TRAIL with ZER or CCB. By contrast, exogenous expression of ATF3 caused a more rapid and elevated expression of DR5, resulting in enhanced sensitivity to apoptotic cell death by TRAIL/ZER or TRAIL/CCB. A reporter assay demonstrated that at least two ATF/cAMP response element motifs as well as C/EBP homologous protein motif at the proximal region of the human DR5 gene promoter were required for ZER-induced DR5 gene transcription. Taken together, our results provide novel insights into the role of ATF3 as an essential transcription factor for p53-independent DR5 induction upon both ZER and CCB treatment, and this may be a useful biomarker for TRAIL-based anticancer therapy.
Keywords: ATF3; Apoptosis; Cancer Therapy; Colon Cancer; DR5; ER Stress; Gene Regulation; Reactive Oxygen Species (ROS); p53-independent.
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Key role of ATF3 in p53-dependent DR5 induction upon DNA damage of human colon cancer cells.Oncogene. 2012 Apr 26;31(17):2210-21. doi: 10.1038/onc.2011.397. Epub 2011 Sep 19. Oncogene. 2012. PMID: 21927023
-
Role of ATF3 in synergistic cancer cell killing by a combination of HDAC inhibitors and agonistic anti-DR5 antibody through ER stress in human colon cancer cells.Biochem Biophys Res Commun. 2014 Mar 7;445(2):320-6. doi: 10.1016/j.bbrc.2014.01.184. Epub 2014 Feb 12. Biochem Biophys Res Commun. 2014. PMID: 24530917
-
Quercetin enhances apoptotic effect of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in ovarian cancer cells through reactive oxygen species (ROS) mediated CCAAT enhancer-binding protein homologous protein (CHOP)-death receptor 5 pathway.Cancer Sci. 2014 May;105(5):520-7. doi: 10.1111/cas.12395. Epub 2014 Apr 11. Cancer Sci. 2014. PMID: 24612139 Free PMC article.
-
Restoring TRAILR2/DR5-Mediated Activation of Apoptosis upon Endoplasmic Reticulum Stress as a Therapeutic Strategy in Cancer.Int J Mol Sci. 2022 Aug 12;23(16):8987. doi: 10.3390/ijms23168987. Int J Mol Sci. 2022. PMID: 36012252 Free PMC article. Review.
-
Regulation of TNF-Related Apoptosis-Inducing Ligand Signaling by Glycosylation.Int J Mol Sci. 2018 Mar 2;19(3):715. doi: 10.3390/ijms19030715. Int J Mol Sci. 2018. PMID: 29498673 Free PMC article. Review.
Cited by
-
Proteotoxic Stress and Cell Death in Cancer Cells.Cancers (Basel). 2020 Aug 23;12(9):2385. doi: 10.3390/cancers12092385. Cancers (Basel). 2020. PMID: 32842524 Free PMC article. Review.
-
Potentiating effect of reovirus on immune checkpoint inhibition in microsatellite stable colorectal cancer.Front Oncol. 2022 Oct 25;12:1018767. doi: 10.3389/fonc.2022.1018767. eCollection 2022. Front Oncol. 2022. PMID: 36387154 Free PMC article.
-
Integrated stress response plasticity governs normal cell adaptation to chronic stress via the PP2A-TFE3-ATF4 pathway.Cell Death Differ. 2024 Dec;31(12):1761-1775. doi: 10.1038/s41418-024-01378-3. Epub 2024 Sep 30. Cell Death Differ. 2024. PMID: 39349971 Free PMC article.
-
Identification of ATF3 as a novel protective signature of quiescent colorectal tumor cells.Cell Death Dis. 2023 Oct 13;14(10):676. doi: 10.1038/s41419-023-06204-1. Cell Death Dis. 2023. PMID: 37833290 Free PMC article.
-
Anti-cancer potential of zerumbone in cancer and glioma: current trends and future perspectives.Med Oncol. 2024 Apr 23;41(5):125. doi: 10.1007/s12032-024-02327-3. Med Oncol. 2024. PMID: 38652207 Review.
References
-
- Hanahan D., Weinberg R. A. (2011) The hallmarks of cancer: the next generation. Cell 144, 646–674 - PubMed
-
- Farczádi E., Szántó J., Kaszás I., Benyó I., Bodnár Z., Szlobodnyik J., Szende B. (1999) Changes in apoptotic and mitotic activity in rectal carcinoma after short-term cytostatic therapy as possible predictive factors. Neoplasma 46, 219–223 - PubMed
-
- Scott N., Hale A., Deakin M., Hand P., Adab F. A., Hall C., Williams G. T., Elder J. B. (1998) A histopathological assessment of the response of rectal adenocarcinoma to combination chemo-radiotherapy: relationship to apoptotic activity, p53 and bcl-2 expression. Eur. J. Surg. Oncol. 24, 169–173 - PubMed
-
- Pan G., Ni J., Wei Y. F., Yu G., Gentz R., Dixit V. M. (1997) An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
