

Transfer RNA and human disease
- PMID: 24917879
- PMCID: PMC4042891
- DOI: 10.3389/fgene.2014.00158
Transfer RNA and human disease
Abstract
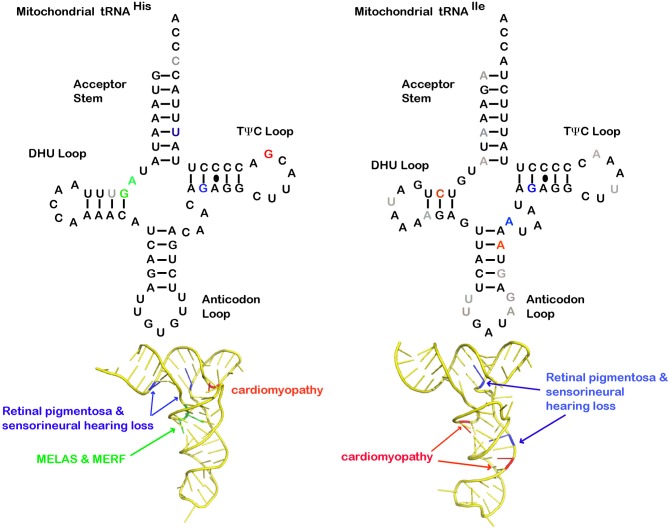
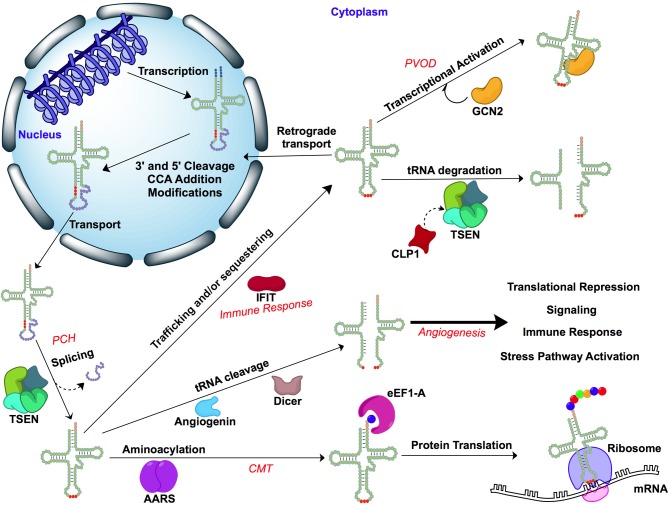
Pathological mutations in tRNA genes and tRNA processing enzymes are numerous and result in very complicated clinical phenotypes. Mitochondrial tRNA (mt-tRNA) genes are "hotspots" for pathological mutations and over 200 mt-tRNA mutations have been linked to various disease states. Often these mutations prevent tRNA aminoacylation. Disrupting this primary function affects protein synthesis and the expression, folding, and function of oxidative phosphorylation enzymes. Mitochondrial tRNA mutations manifest in a wide panoply of diseases related to cellular energetics, including COX deficiency (cytochrome C oxidase), mitochondrial myopathy, MERRF (Myoclonic Epilepsy with Ragged Red Fibers), and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes). Diseases caused by mt-tRNA mutations can also affect very specific tissue types, as in the case of neurosensory non-syndromic hearing loss and pigmentary retinopathy, diabetes mellitus, and hypertrophic cardiomyopathy. Importantly, mitochondrial heteroplasmy plays a role in disease severity and age of onset as well. Not surprisingly, mutations in enzymes that modify cytoplasmic and mitochondrial tRNAs are also linked to a diverse range of clinical phenotypes. In addition to compromised aminoacylation of the tRNAs, mutated modifying enzymes can also impact tRNA expression and abundance, tRNA modifications, tRNA folding, and even tRNA maturation (e.g., splicing). Some of these pathological mutations in tRNAs and processing enzymes are likely to affect non-canonical tRNA functions, and contribute to the diseases without significantly impacting on translation. This chapter will review recent literature on the relation of mitochondrial and cytoplasmic tRNA, and enzymes that process tRNAs, to human disease. We explore the mechanisms involved in the clinical presentation of these various diseases with an emphasis on neurological disease.
Keywords: Usher syndrome Type IIIB; aminoacyl-tRNA synthetase; localized translation; mitochondrial disease; neurodegenerative disease; tRNA.
Figures
Similar articles
-
Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes.Mol Cell Biol. 2010 May;30(9):2147-54. doi: 10.1128/MCB.01614-09. Epub 2010 Mar 1. Mol Cell Biol. 2010. PMID: 20194621 Free PMC article.
-
Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease.Front Genet. 2015 Mar 10;6:79. doi: 10.3389/fgene.2015.00079. eCollection 2015. Front Genet. 2015. PMID: 25806043 Free PMC article. Review.
-
Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria.Nucleic Acids Res. 2011 Oct;39(18):8173-86. doi: 10.1093/nar/gkr546. Epub 2011 Jun 30. Nucleic Acids Res. 2011. PMID: 21724600 Free PMC article.
-
Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases.Annu Rev Genet. 2011;45:299-329. doi: 10.1146/annurev-genet-110410-132531. Epub 2011 Sep 6. Annu Rev Genet. 2011. PMID: 21910628 Review.
-
Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs.Wiley Interdiscip Rev RNA. 2011 May-Jun;2(3):376-86. doi: 10.1002/wrna.65. Epub 2011 Feb 25. Wiley Interdiscip Rev RNA. 2011. PMID: 21957023 Review.
Cited by
-
Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation.Nat Commun. 2020 Aug 14;11(1):4104. doi: 10.1038/s41467-020-17879-x. Nat Commun. 2020. PMID: 32796835 Free PMC article.
-
The fitness landscape of a tRNA gene.Science. 2016 May 13;352(6287):837-40. doi: 10.1126/science.aae0568. Epub 2016 Apr 14. Science. 2016. PMID: 27080104 Free PMC article.
-
Pathways to disease from natural variations in human cytoplasmic tRNAs.J Biol Chem. 2019 Apr 5;294(14):5294-5308. doi: 10.1074/jbc.REV118.002982. Epub 2019 Jan 14. J Biol Chem. 2019. PMID: 30643023 Free PMC article. Review.
-
Impaired Bioenergetics in Mutant Mitochondrial DNA Determines Cell Fate During Seizure-Like Activity.Mol Neurobiol. 2019 Jan;56(1):321-334. doi: 10.1007/s12035-018-1078-9. Epub 2018 Apr 27. Mol Neurobiol. 2019. PMID: 29704197
-
Recessive VARS2 mutation underlies a novel syndrome with epilepsy, mental retardation, short stature, growth hormone deficiency, and hypogonadism.Hum Genomics. 2017 Nov 14;11(1):28. doi: 10.1186/s40246-017-0124-4. Hum Genomics. 2017. PMID: 29137650 Free PMC article.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources