

RIPK1- and RIPK3-induced cell death mode is determined by target availability
- PMID: 24902899
- PMCID: PMC4158685
- DOI: 10.1038/cdd.2014.70
RIPK1- and RIPK3-induced cell death mode is determined by target availability
Abstract
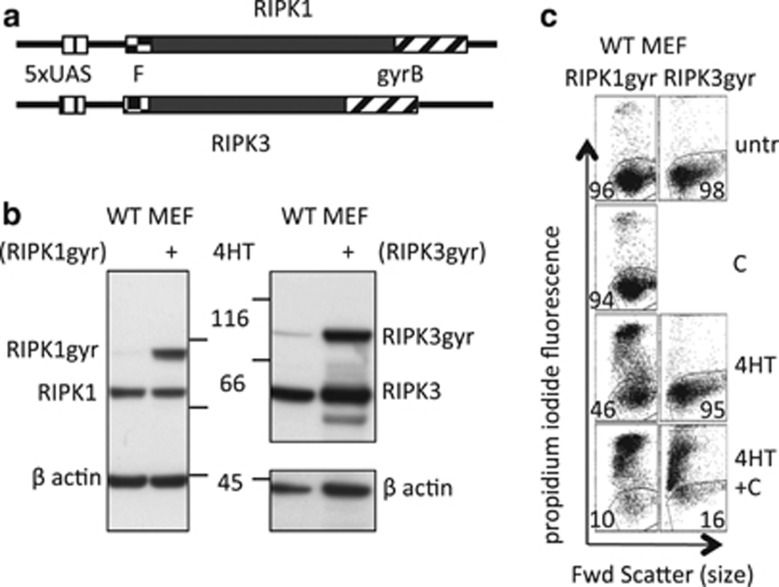
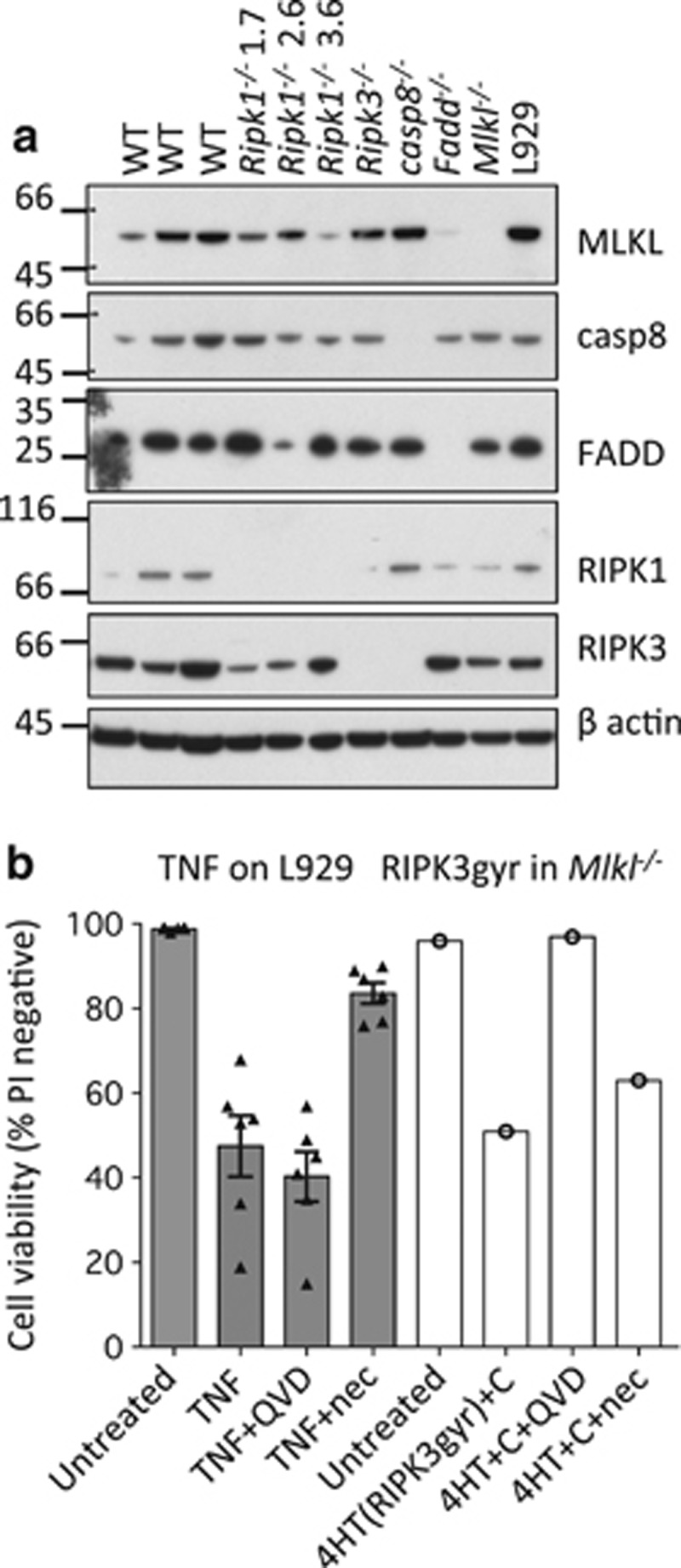
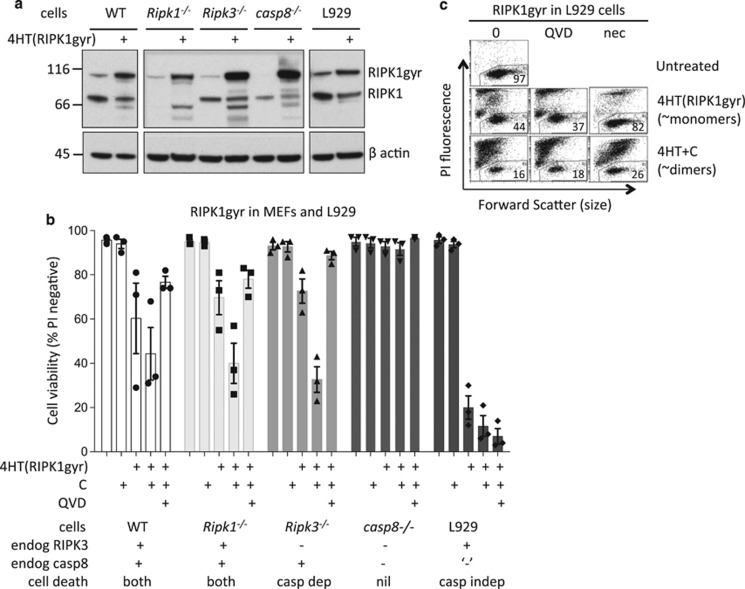
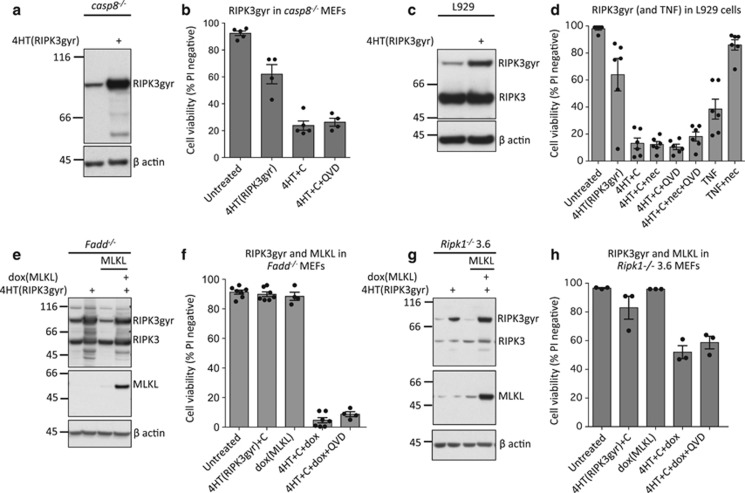
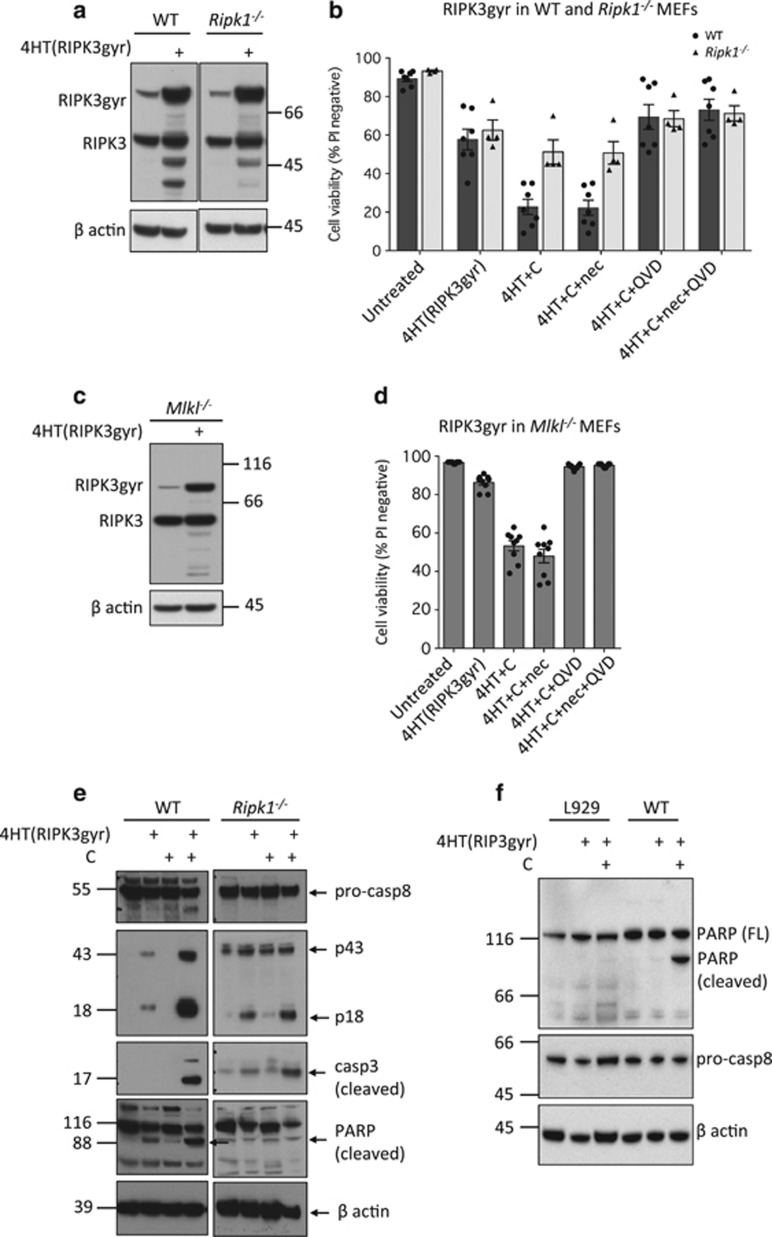
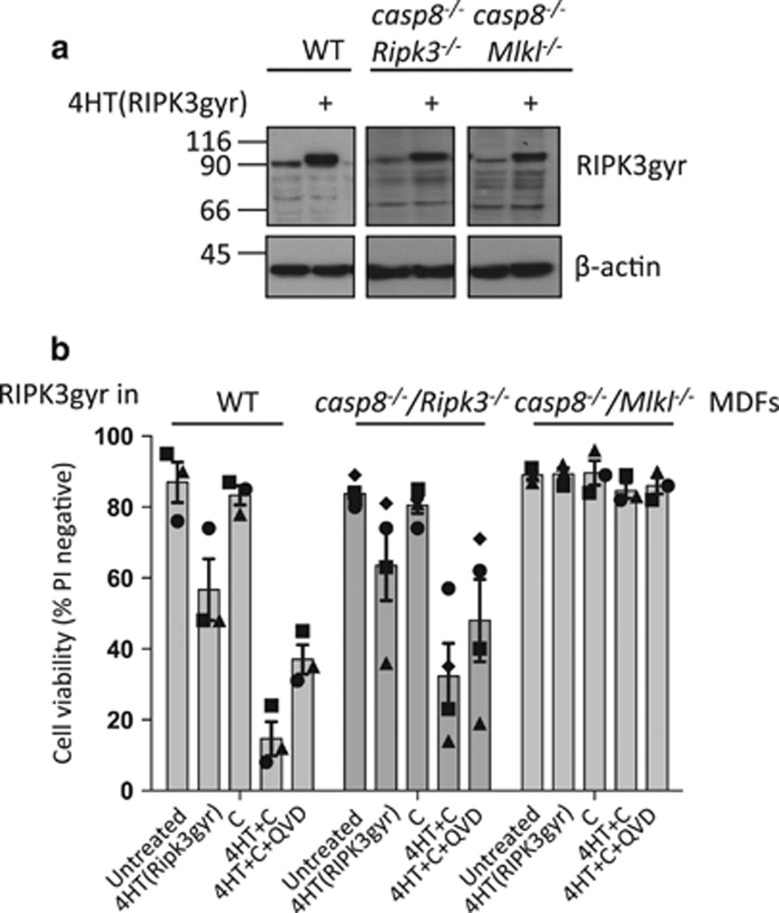
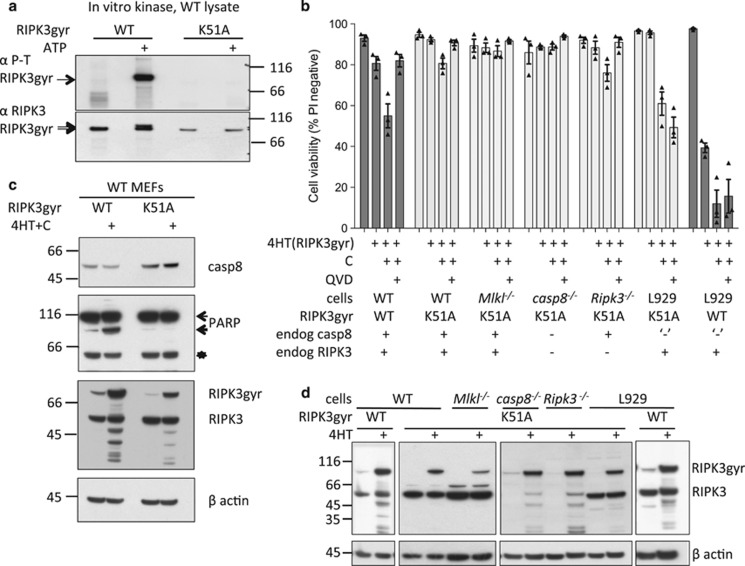
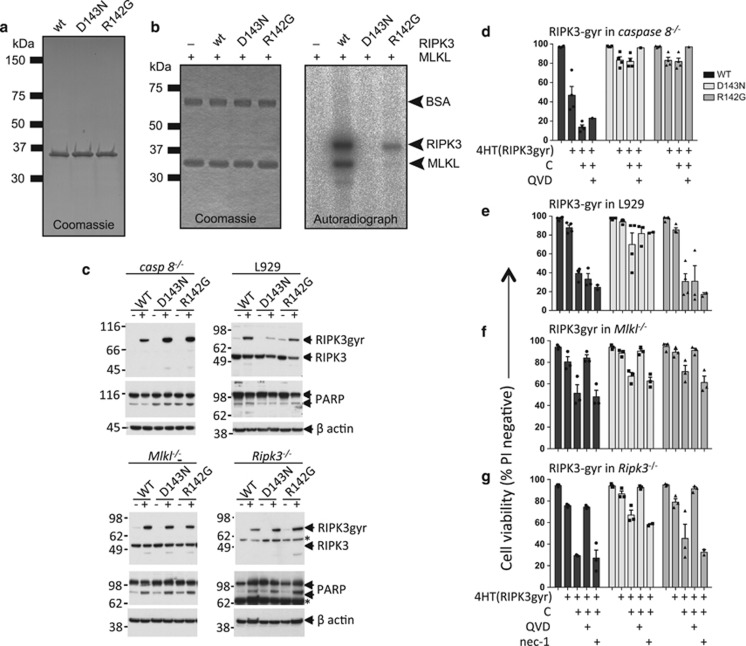
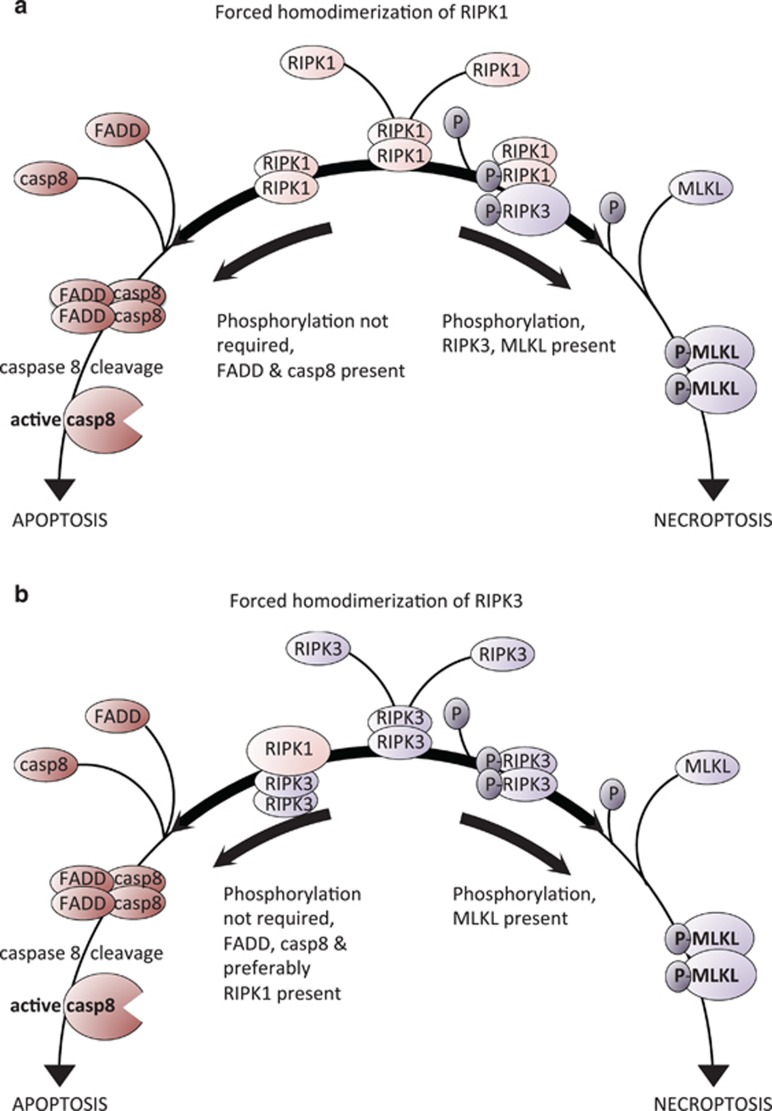
Both receptor-interacting protein kinase 1 (RIPK1) and RIPK3 can signal cell death following death receptor ligation. To study the requirements for RIPK-triggered cell death in the absence of death receptor signaling, we engineered inducible versions of RIPK1 and RIPK3 that can be activated by dimerization with the antibiotic coumermycin. In the absence of TNF or other death ligands, expression and dimerization of RIPK1 was sufficient to cause cell death by caspase- or RIPK3-dependent mechanisms. Dimerized RIPK3 induced cell death by an MLKL-dependent mechanism but, surprisingly, also induced death mediated by FADD, caspase 8 and RIPK1. Catalytically active RIPK3 kinase domains were essential for MLKL-dependent but not for caspase 8-dependent death. When RIPK1 or RIPK3 proteins were dimerized, the mode of cell death was determined by the availability of downstream molecules such as FADD, caspase 8 and MLKL. These observations imply that rather than a 'switch' operating between the two modes of cell death, the final mechanism depends on levels of the respective signaling and effector proteins.
Figures
Similar articles
-
RIPK1 prevents TRADD-driven, but TNFR1 independent, apoptosis during development.Cell Death Differ. 2019 May;26(5):877-889. doi: 10.1038/s41418-018-0166-8. Epub 2018 Sep 5. Cell Death Differ. 2019. PMID: 30185824 Free PMC article.
-
RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis.Cell Death Differ. 2014 Oct;21(10):1511-21. doi: 10.1038/cdd.2014.76. Epub 2014 Jun 6. Cell Death Differ. 2014. PMID: 24902904 Free PMC article.
-
RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3.Cell. 2014 May 22;157(5):1189-202. doi: 10.1016/j.cell.2014.04.018. Epub 2014 May 8. Cell. 2014. PMID: 24813850 Free PMC article.
-
RIPK-dependent necrosis and its regulation by caspases: a mystery in five acts.Mol Cell. 2011 Oct 7;44(1):9-16. doi: 10.1016/j.molcel.2011.09.003. Mol Cell. 2011. PMID: 21981915 Free PMC article. Review.
-
Necroptotic Cell Death Signaling and Execution Pathway: Lessons from Knockout Mice.Mediators Inflamm. 2015;2015:128076. doi: 10.1155/2015/128076. Epub 2015 Sep 27. Mediators Inflamm. 2015. PMID: 26491219 Free PMC article. Review.
Cited by
-
DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death.Cell Host Microbe. 2016 Nov 9;20(5):674-681. doi: 10.1016/j.chom.2016.09.014. Epub 2016 Oct 13. Cell Host Microbe. 2016. PMID: 27746097 Free PMC article.
-
Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics.Cell Res. 2015 Jun;25(6):707-25. doi: 10.1038/cr.2015.56. Epub 2015 May 8. Cell Res. 2015. PMID: 25952668 Free PMC article.
-
P2X7 Receptor-Associated Programmed Cell Death in the Pathophysiology of Hemorrhagic Stroke.Curr Neuropharmacol. 2018;16(9):1282-1295. doi: 10.2174/1570159X16666180516094500. Curr Neuropharmacol. 2018. PMID: 29766811 Free PMC article. Review.
-
Triad3a induces the degradation of early necrosome to limit RipK1-dependent cytokine production and necroptosis.Cell Death Dis. 2018 May 22;9(6):592. doi: 10.1038/s41419-018-0672-0. Cell Death Dis. 2018. PMID: 29789521 Free PMC article.
-
A toolbox for imaging RIPK1, RIPK3, and MLKL in mouse and human cells.Cell Death Differ. 2021 Jul;28(7):2126-2144. doi: 10.1038/s41418-021-00742-x. Epub 2021 Feb 15. Cell Death Differ. 2021. PMID: 33589776 Free PMC article.
References
-
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. - PubMed
-
- Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. - PubMed
-
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. - PubMed
-
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. - PubMed
-
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
