

Prediction and redesign of protein-protein interactions
- PMID: 24878423
- PMCID: PMC4246023
- DOI: 10.1016/j.pbiomolbio.2014.05.004
Prediction and redesign of protein-protein interactions
Abstract
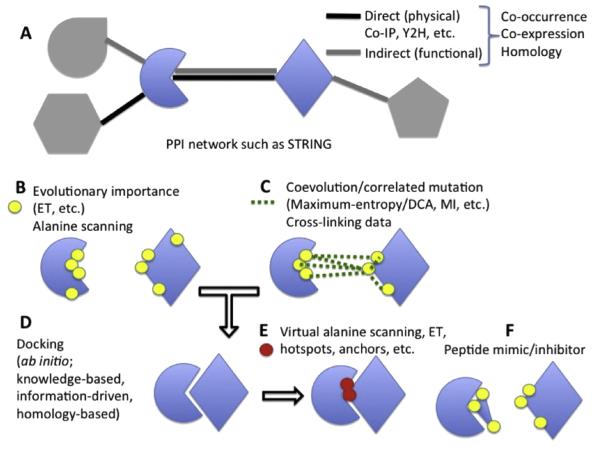
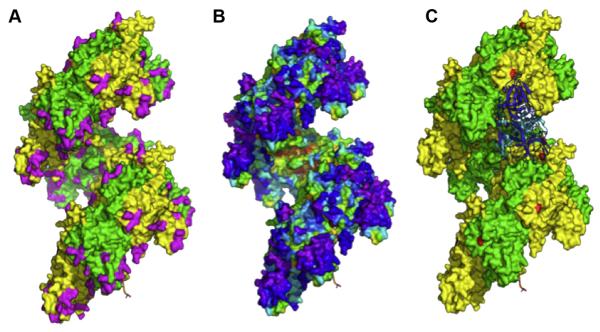
Understanding the molecular basis of protein function remains a central goal of biology, with the hope to elucidate the role of human genes in health and in disease, and to rationally design therapies through targeted molecular perturbations. We review here some of the computational techniques and resources available for characterizing a critical aspect of protein function - those mediated by protein-protein interactions (PPI). We describe several applications and recent successes of the Evolutionary Trace (ET) in identifying molecular events and shapes that underlie protein function and specificity in both eukaryotes and prokaryotes. ET is a part of analytical approaches based on the successes and failures of evolution that enable the rational control of PPI.
Keywords: Evolutionary trace; Functional annotation; Functional sites; Molecular evolution; Protein design; Protein–protein interaction networks.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Ultra-fast FFT protein docking on graphics processors.Bioinformatics. 2010 Oct 1;26(19):2398-405. doi: 10.1093/bioinformatics/btq444. Epub 2010 Aug 4. Bioinformatics. 2010. PMID: 20685958
-
Flexible Backbone Methods for Predicting and Designing Peptide Specificity.Methods Mol Biol. 2017;1561:173-187. doi: 10.1007/978-1-4939-6798-8_10. Methods Mol Biol. 2017. PMID: 28236238
-
Computational structural analysis of protein interactions and networks.Proteomics. 2012 May;12(10):1697-705. doi: 10.1002/pmic.201100597. Proteomics. 2012. PMID: 22593000 Review.
-
Designing specific protein-protein interactions using computation, experimental library screening, or integrated methods.Protein Sci. 2012 Jul;21(7):949-63. doi: 10.1002/pro.2096. Epub 2012 Jun 8. Protein Sci. 2012. PMID: 22593041 Free PMC article. Review.
-
Systematic computational prediction of protein interaction networks.Phys Biol. 2011 Jun;8(3):035008. doi: 10.1088/1478-3975/8/3/035008. Epub 2011 May 13. Phys Biol. 2011. PMID: 21572181
Cited by
-
Paradoxes of early stages of evolution of life and biological complexity.Orig Life Evol Biosph. 2015 Jun;45(1-2):163-71. doi: 10.1007/s11084-015-9414-9. Epub 2015 Mar 11. Orig Life Evol Biosph. 2015. PMID: 25754592
-
Application of docking methodologies to modeled proteins.Proteins. 2020 Sep;88(9):1180-1188. doi: 10.1002/prot.25889. Epub 2020 Mar 20. Proteins. 2020. PMID: 32170770 Free PMC article.
-
Structural Perspectives on the Evolutionary Expansion of Unique Protein-Protein Binding Sites.Biophys J. 2015 Sep 15;109(6):1295-306. doi: 10.1016/j.bpj.2015.06.056. Epub 2015 Jul 23. Biophys J. 2015. PMID: 26213149 Free PMC article.
-
A hybrid method for protein-protein interface prediction.Protein Sci. 2016 Jan;25(1):159-65. doi: 10.1002/pro.2744. Epub 2015 Jul 21. Protein Sci. 2016. PMID: 26178156 Free PMC article.
-
Challenges in structural approaches to cell modeling.J Mol Biol. 2016 Jul 31;428(15):2943-64. doi: 10.1016/j.jmb.2016.05.024. Epub 2016 May 30. J Mol Biol. 2016. PMID: 27255863 Free PMC article. Review.
References
-
- Aloy P, Russell RB. Structural systems biology: modelling protein interactions. Nat. Rev. Mol. Cell. Biol. 2006;7:188–197. - PubMed
-
- Aloy P, Querol E, Aviles FX, Sternberg MJ. Automated structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J. Mol. Biol. 2001;311:395–408. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
