

Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm
- PMID: 24812670
- PMCID: PMC4038579
- DOI: 10.1172/JCI74182
Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm
Abstract
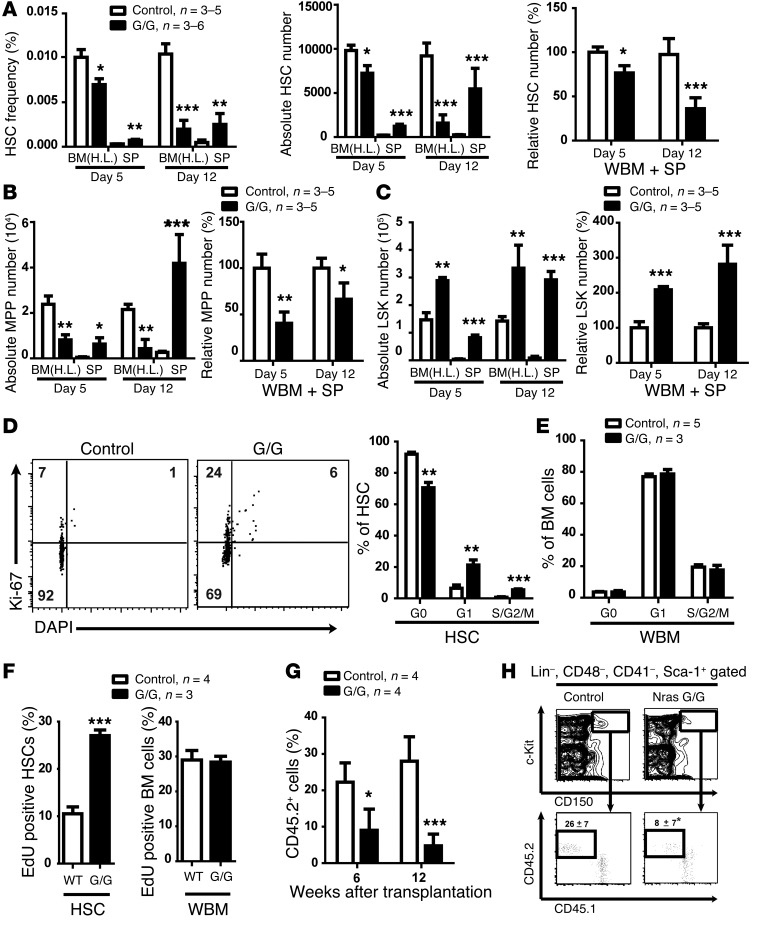
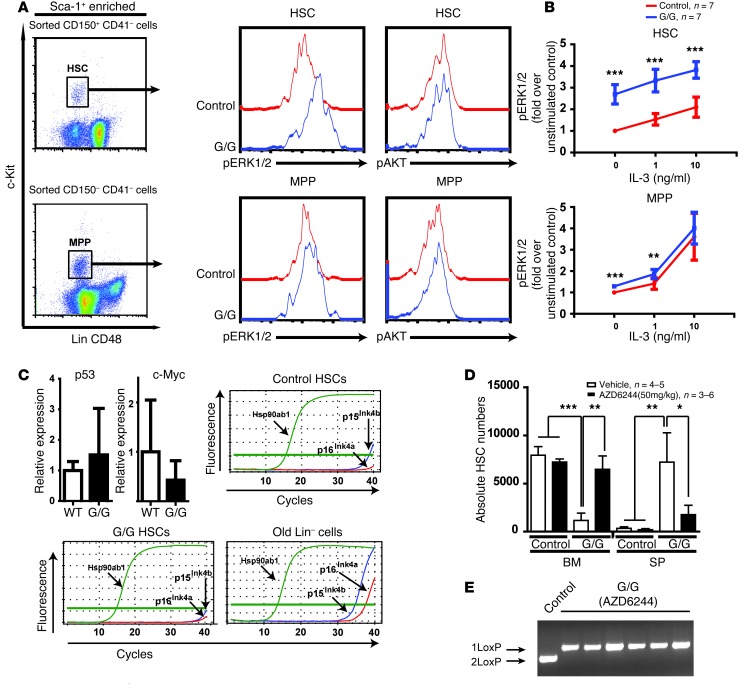
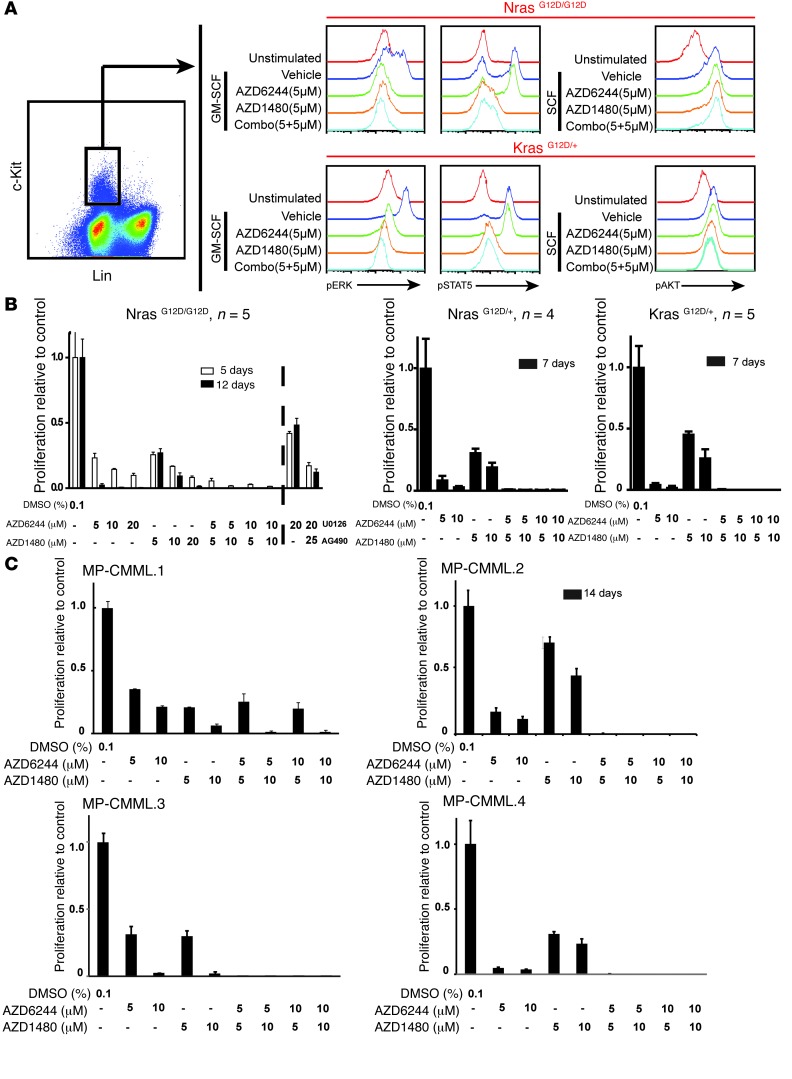
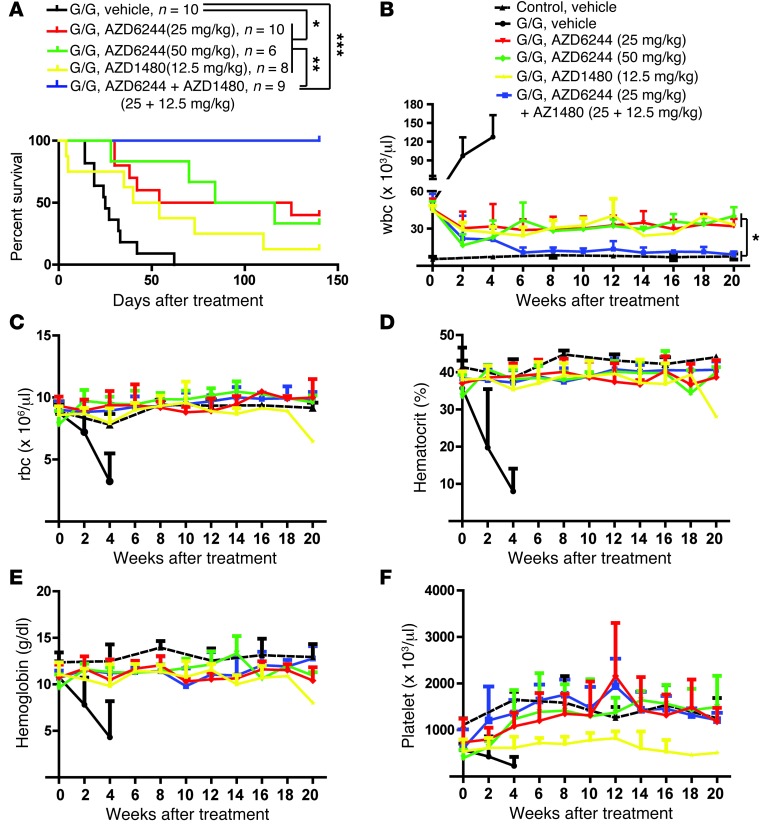
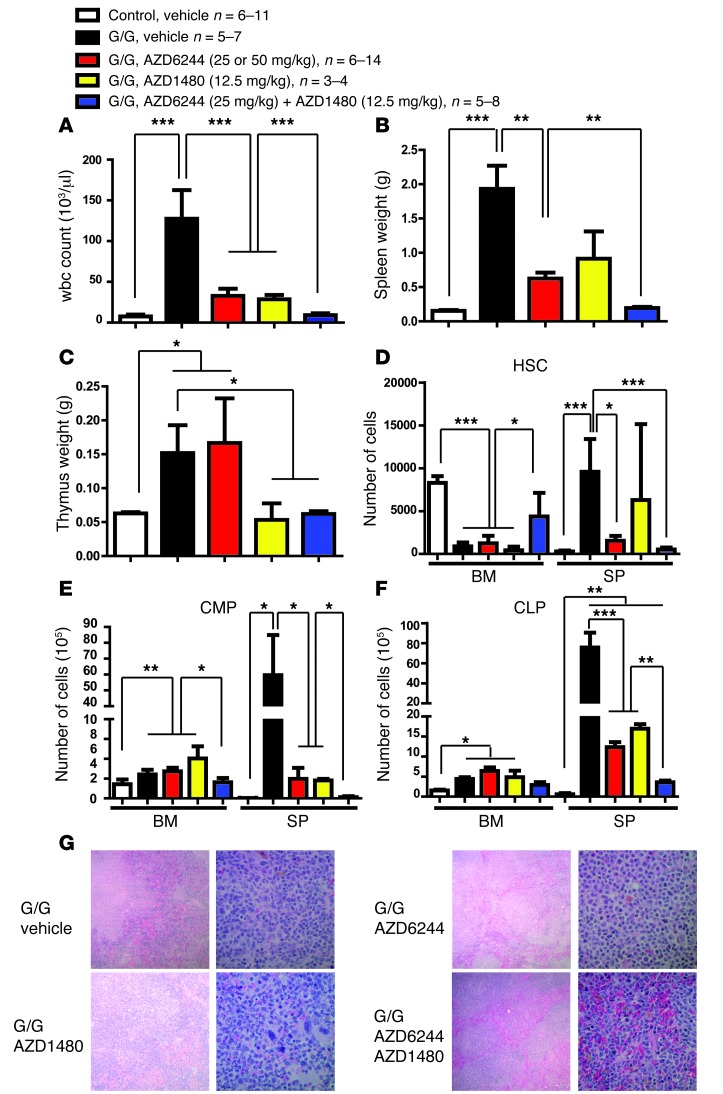
Overactive RAS signaling is prevalent in juvenile myelomonocytic leukemia (JMML) and the myeloproliferative variant of chronic myelomonocytic leukemia (MP-CMML) in humans, and both are refractory to conventional chemotherapy. Conditional activation of a constitutively active oncogenic Nras (NrasG12D/G12D) in murine hematopoietic cells promotes an acute myeloproliferative neoplasm (MPN) that recapitulates many features of JMML and MP-CMML. We found that NrasG12D/G12D-expressing HSCs, which serve as JMML/MP-CMML-initiating cells, show strong hyperactivation of ERK1/2, promoting hyperproliferation and depletion of HSCs and expansion of downstream progenitors. Inhibition of the MEK pathway alone prolonged the presence of NrasG12D/G12D-expressing HSCs but failed to restore their proper function. Consequently, approximately 60% of NrasG12D/G12D mice treated with MEK inhibitor alone died within 20 weeks, and the remaining animals continued to display JMML/MP-CMML-like phenotypes. In contrast, combined inhibition of MEK and JAK/STAT signaling, which is commonly hyperactivated in human and mouse CMML, potently inhibited human and mouse CMML cell growth in vitro, rescued mutant NrasG12D/G12D-expressing HSC function in vivo, and promoted long-term survival without evident disease manifestation in NrasG12D/G12D animals. These results provide a strong rationale for further exploration of combined targeting of MEK/ERK and JAK/STAT in treating patients with JMML and MP-CMML.
Figures

Similar articles
-
Deficiency of β Common Receptor Moderately Attenuates the Progression of Myeloproliferative Neoplasm in NrasG12D/+ Mice.J Biol Chem. 2015 Jul 31;290(31):19093-103. doi: 10.1074/jbc.M115.653154. Epub 2015 Jun 16. J Biol Chem. 2015. PMID: 26082490 Free PMC article.
-
A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice.Sci Transl Med. 2011 Mar 30;3(76):76ra27. doi: 10.1126/scitranslmed.3001069. Sci Transl Med. 2011. PMID: 21451123 Free PMC article.
-
Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice.J Clin Invest. 2013 Jan;123(1):335-9. doi: 10.1172/JCI63193. Epub 2012 Dec 10. J Clin Invest. 2013. PMID: 23221337 Free PMC article.
-
Treatment advances for pediatric and adult onset neoplasms with monocytosis.Curr Hematol Malig Rep. 2021 Jun;16(3):256-266. doi: 10.1007/s11899-021-00622-8. Epub 2021 Mar 16. Curr Hematol Malig Rep. 2021. PMID: 33728588 Review.
-
Chronic myelomonocytic leukemia: myeloproliferative variant.Curr Hematol Rep. 2004 May;3(3):218-26. Curr Hematol Rep. 2004. PMID: 15087071 Review.
Cited by
-
MEK inhibitors for neurofibromatosis type 1 manifestations: Clinical evidence and consensus.Neuro Oncol. 2022 Nov 2;24(11):1845-1856. doi: 10.1093/neuonc/noac165. Neuro Oncol. 2022. PMID: 35788692 Free PMC article. Review.
-
Single agent BMS-911543 Jak2 inhibitor has distinct inhibitory effects on STAT5 signaling in genetically engineered mice with pancreatic cancer.Oncotarget. 2015 Dec 29;6(42):44509-22. doi: 10.18632/oncotarget.6332. Oncotarget. 2015. PMID: 26575024 Free PMC article.
-
Novel Therapies for Myelofibrosis.Curr Hematol Malig Rep. 2017 Dec;12(6):611-624. doi: 10.1007/s11899-017-0403-0. Curr Hematol Malig Rep. 2017. PMID: 29098608 Free PMC article. Review.
-
Deletion of Ptpn1 induces myeloproliferative neoplasm.Leukemia. 2017 May;31(5):1229-1234. doi: 10.1038/leu.2017.31. Epub 2017 Jan 23. Leukemia. 2017. PMID: 28111468 Free PMC article. No abstract available.
-
Molecular Pathogenesis of Chronic Myelomonocytic Leukemia and Potential Molecular Targets for Treatment Approaches.Front Oncol. 2021 Sep 30;11:751668. doi: 10.3389/fonc.2021.751668. eCollection 2021. Front Oncol. 2021. PMID: 34660314 Free PMC article.
References
-
- Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia. 2008;22(7):1335–1342. - PubMed
-
- Dunbar AJ, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68(24):10349–10357. doi: 10.1158/0008-5472.CAN-08-2754. - DOI - PMC - PubMed
-
- Kohlmann A, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28(24):3858–3865. doi: 10.1200/JCO.2009.27.1361. - DOI - PubMed
-
- Kato M, et al. Aggressive transformation of juvenile myelomonocytic leukemia associated with duplication of oncogenic KRAS due to acquired uniparental disomy. J Pediatr. 2013;162(6):1285–1288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
