

Review
doi: 10.1021/cr400525m.
Epub 2014 Apr 29.
Classification of intrinsically disordered regions and proteins
Affiliations
- PMID: 24773235
- PMCID: PMC4095912
- DOI: 10.1021/cr400525m
Item in Clipboard
Review
Classification of intrinsically disordered regions and proteins
Chem Rev.
.
No abstract available
Figures
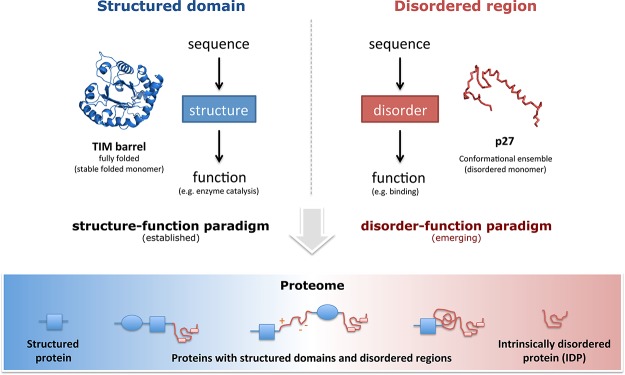
Structured domains and intrinsically disordered regions
(IDRs)
are two fundamental classes of functional building blocks of proteins.
The synergy between disordered regions and structured domains increases
the functional versatility of proteins. Adapted with permission from
ref (50). Copyright
2012 American Association for the Advancement of Science.
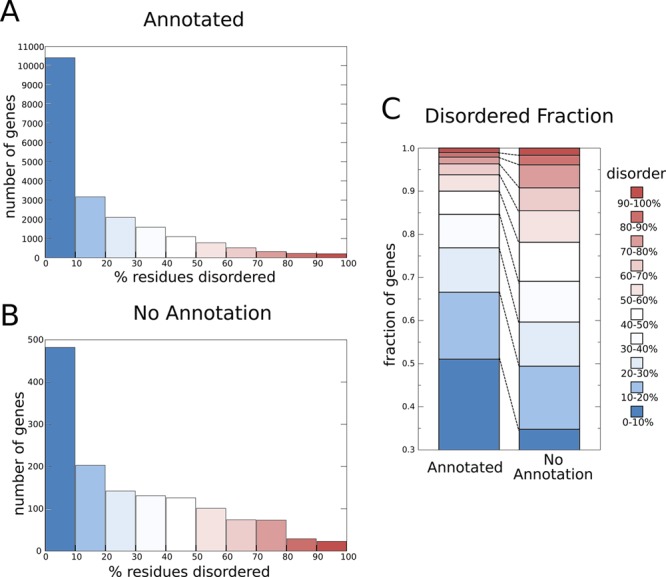
The number of protein-coding genes in the human genome with various
amounts of disorder. Histograms of the numbers of human genes with
annotation (A) and without annotation (B), grouped by the percentage
of disordered residues. (C) A comparison of the fraction of annotated
and unannotated human genes with different amounts of disorder. Residues
in each protein are defined as disordered when there is a consensus
between >75% of the predictors in the D2P2 database at that position. The set
of human genes was
taken from Ensembl release 63, and the
representative protein coded for by the longest transcript was used
in each case. The annotation was taken from the description field
with “open reading frame”, “hypothetical”,
“uncharacterized”, and “putative protein”
treated as no annotation.
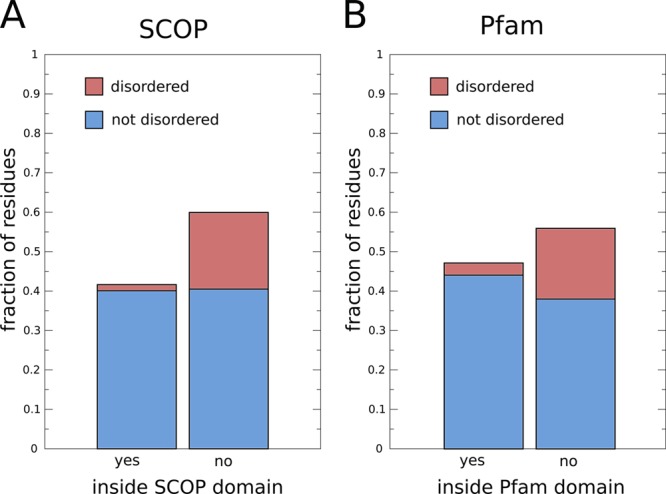
The fraction of disordered residues located in domains in human
protein-coding genes: (A) residues inside (left) and outside (right)
of SCOP domains, and (B) residues inside
(left) and outside (right) of Pfam domains (only curated Pfam domains
were considered, i.e., Pfam-A). The SCOP
domains in human proteins are defined by the SUPERFAMILY database. Disordered residues were taken from the D2P2 database (when
there is a consensus between >75% of the disorder predictors).
The
set of human genes was taken from Ensembl release 63.
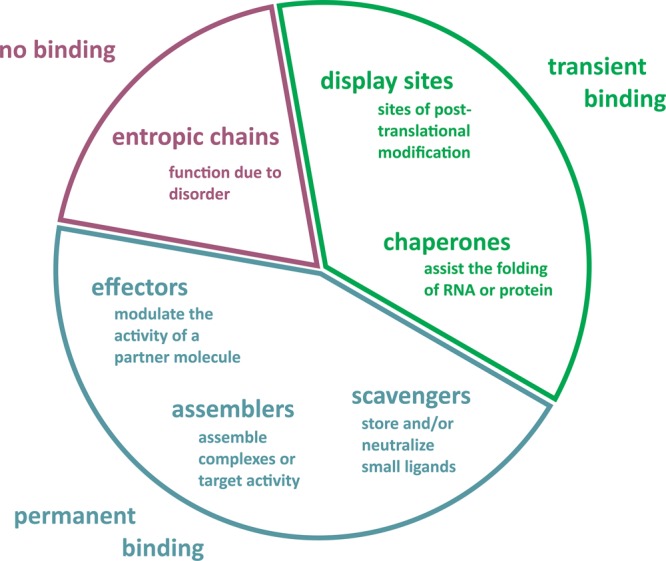
Functional classification scheme of IDRs.
The function of disordered
regions can stem directly from their highly flexible nature, when
they fulfill entropic chain functions (such as linkers and spacers,
indicated in dark-tone red), or from their ability to bind to partner
molecules (proteins, other macromolecules, or small molecules). In
the latter case, they bind either transiently as display sites of
post-translational modifications or as chaperones (indicated in green),
or they bind permanently as effectors, assemblers, or scavengers (indicated
in dark-tone blue). More extensive descriptions and examples are found
in the main text. Adapted with permission from ref (58). Copyright 2005 Elsevier.
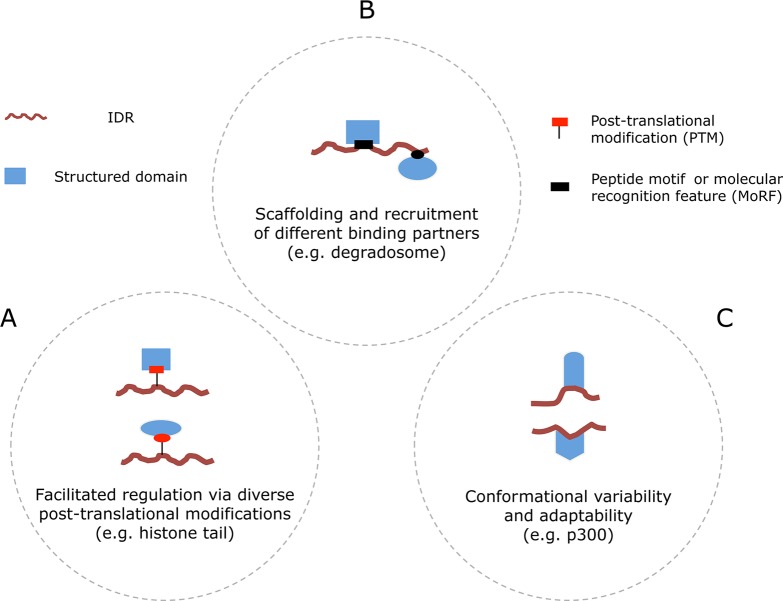
Functional classification of IDRs according
to their interaction
features. (A) The flexibility of IDRs facilitates access to enzymes
that catalyze post-translational modifications and effectors that
bind these PTMs. This permits combinatorial regulation and reuse of
the same components in multiple biological processes. (B) The availability
of molecular recognition features and linear motifs within the IDRs
enables the fishing for (“fly casting”) and gathering
of different partners. (C) Conformational variability enables a nearly
perfect molding to fit the binding interfaces of very diverse interaction
partners. Context-dependent folding of an IDR can activate signaling
processes in one case or inhibit them in another, resulting in completely
different outcomes. Adapted with permission from ref (39). Copyright 2009 Elsevier.
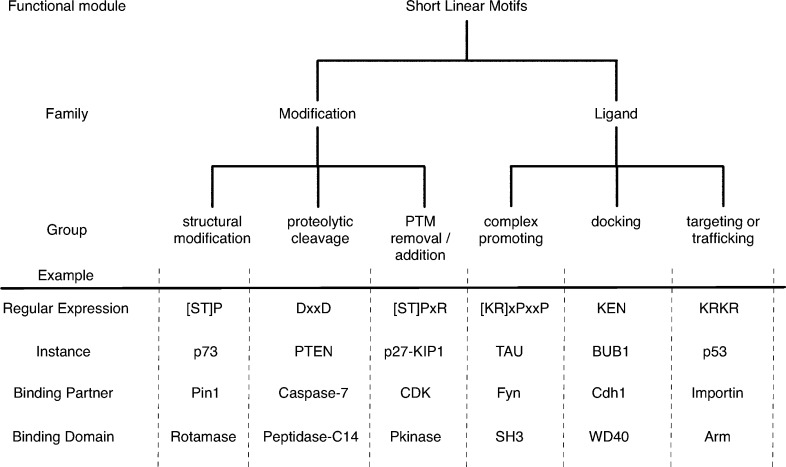
Functional classification of linear motifs. Linear motifs can be
divided into two major families, which each have three further subgroups.
The modification class motifs all act as recognition sites for enzyme
active sites, whereas the ligand class motifs are always recognized
by the binding surface of a protein partner. More detailed classification
beyond the graph shown here is possible. For example, an important
subgroup of docking motifs are the degrons, which regulate protein
stability by recruiting members of the ubiquitin–proteasome
system. In the regular expressions, x corresponds to any amino acid,
while other letters represent single letter codes of amino acids;
letters within square brackets mean either residue is allowed in that
position.
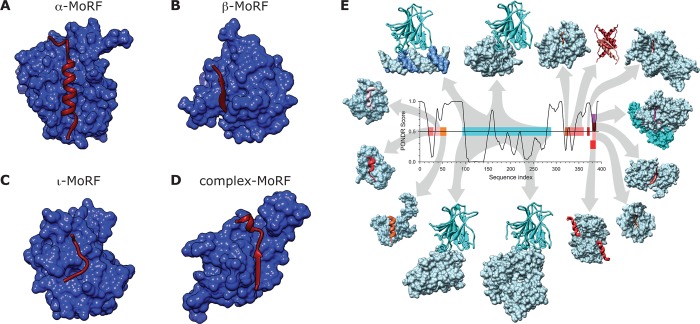
Classification of molecular recognition
features (MoRFs) based
on the secondary structure of the bound state. MoRFs (red ribbons)
undergo disorder-to-order transition upon binding their partners (blue
surfaces). (A) α-MoRF. BH3 domain of BAD (MoRF) bound to bcl-xl
(partner) (PDB ID: 1G5J). (B) β-MoRF. Inhibitor of apoptosis protein DIAP1 (partner)
bound to N-terminus of cell death protein GRIM (MoRF) (PDB ID: 1JD5). (C) ι-MoRF.
AP-2 (partner) bound to the recognition motif of amphiphysin (MoRF)
(PDB ID: 1KY7). (D) Complex-MoRF. Phosphotyrosine-binding domain (PTB) of the
X11 protein (partner) bound to amyloid β A4 protein (MoRF) (PDB
ID: 1X11). Note
that the PTB domain of X11 actually binds unphosphorylated peptides
and is a PTB by sequence similarity. Panels A–D reprinted with
permission from ref (122). Copyright 2007 American Chemical Society. (E) Promiscuity of disorder-controlled
interactions illustrated by the p53 interaction network. A structure
versus disorder prediction on the p53 amino acid sequence is shown
in the center of the figure (up = disorder, down = order) along with
the structures of various regions of p53 bound to 14 different partners.
The predictions for a central region of structure, and the disordered
amino and carbonyl termini have been confirmed experimentally for
p53. The various regions of p53 are color coded to show their structures
in the complex and to map the binding segments to the amino acid sequence.
Starting with the p53–DNA complex (top, left, magenta protein,
blue DNA), and moving in a clockwise direction, the Protein Data Bank IDs and partner names are given as follows
for the 14 complexes: (1tsr – DNA), (1gzh – 53BP1), (1q2d – gcn5),
(3sak –
p53 (tetramerization domain)), (1xqh – set9), (1h26 – cyclin
A), (1ma3 –
sirtuin), (1jsp – CBP bromo domain), (1dt7 – s100ββ), (2h1l – sv40 Large
T antigen), (1ycs – 53BP2), (2gs0 – PH), (1ycr – MDM2), and (2b3g – RPA70). Reprinted with permission from ref (40). Copyright 2010 Elsevier.
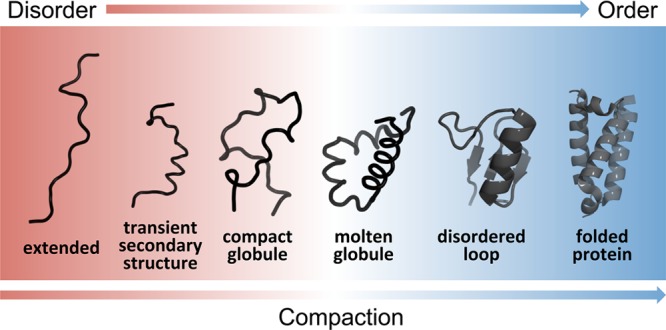
Schematic representation of the continuum
model of protein structure.
The color gradient represents a continuum of conformational states
ranging from highly dynamic, expanded conformational ensembles (red)
to compact, dynamically restricted, fully folded globular states (blue).
Dynamically disordered states are represented by heavy lines, stably
folded structures as cartoons. A characteristic of IDPs is that they
rapidly interconvert between multiple states in the dynamic conformational
ensemble. In the continuum model, the proteome would populate the
entire spectrum of dynamics, disorder, and folded structure depicted.
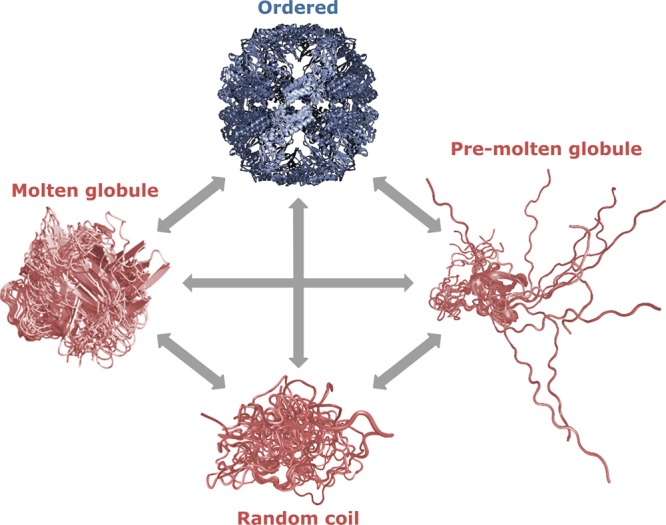
The protein quartet model of protein conformational states. In
accordance with this model, protein function arises from four types
of conformations of the polypeptide chain (ordered forms, molten globules,
pre-molten globules, and random coils) and transitions between any
of these states.
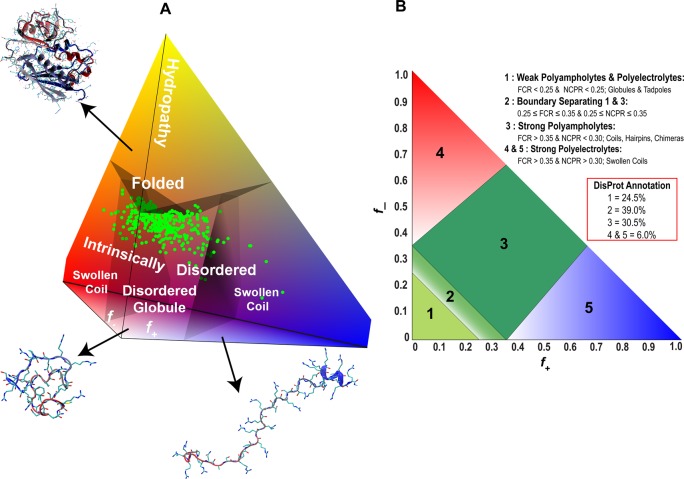
Original and modified diagram-of-states
to classify predicted conformational
properties of IDPs (and IDRs modeled as IDPs). (A) The original diagram
predicts that sequences with a net charge per residue above 0.25 will
be swollen coils. The three axes denote the fraction of positively
charged residues, f+, the fraction of
negatively charged residues, f–, and the hydropathy. All three parameters are calculated from the
amino acid composition. Green dots correspond to 364 curated disordered
sequences extracted from the DisProt database. These sequences have hydropathy values that designate them
as being disordered; that is, they lie in the bottom portion of the
pyramid by definition. Additional filters were used for chain length
(more than 30 residues) and the fraction of proline residues (fpro < 0.3). 97% of sequences used in this
annotation have a net charge per residue of less than 0.26 and are
thus predicted to be globule formers. Adapted from ref (166). Copyright 2010 National Academy of Sciences of the United States
of America. (B) Modified diagram-of-states from panel (A) with a focus
only on the bottom portion of the pyramid (i.e., stipulating that
the hydropathy is low enough to be ignored). The polyampholytic contribution expands the space encompassed by
nonglobule-formers by subdividing the disordered globules space in
panel (A) into three distinct regions of which sequences in regions
2 and 3 actually may not form globules. In these polyampholytic regions,
one has to account for the total charge, in terms of the fraction
of charged residues (FCR), as well as the net charge per residue (NCPR)
as opposed to NCPR alone. Conformations in regions 2 and 3 are expected
to be random-coil-like if oppositely charged residues are well mixed
in the linear sequence. Otherwise, one can expect compact or semicompact
conformations. The classification scheme uses only the amino acid
sequence as input. Reprinted with permission from ref (204). Copyright 2013 National
Academy of Sciences of the United States of America.
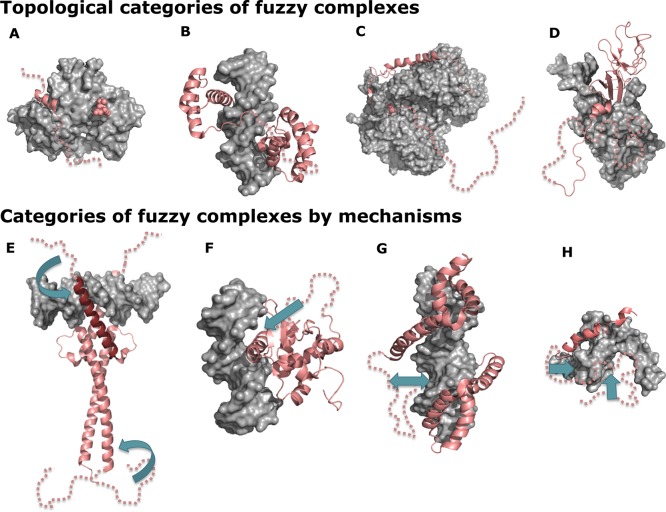
Classification of fuzzy complexes by topology (upper panel) and
by mechanism (lower panel). Blue arrows indicate interactions between
fuzzy disordered regions and structured molecules. Protein Data Bank identifiers for the structures are given in
parentheses. Topological categories: (A) Polymorphic. The WH2 domain
of ciboulot interacts with actin in alternative locations: via an
18-residue segment (3u9z) or via only three residues (2ff3). The flanking regions remain dynamically
disordered. (B) Clamp. The Oct-1 transcription factor has a bipartite
DNA recognition motif. The two globular binding domains are connected
by a 23 residue long disordered linker (1hf0), shortening of which reduces binding
affinity. (C) Flanking. The p27Kip1 cell-cycle kinase inhibitor
binds to the cyclin–Cdk2 complex (1jsu). The kinase binding site is flanked
by a ∼100 residue long disordered linker, which enables T187
at the C-terminus to be phosphorylated. (D) Random. UmuD2 is a dimer
that is produced from UmuD by RecA-facilitated self-cleavage (1i4v). The resulting
proteins exhibit a random coil signal in circular dichroism experiments
at physiologically relevant concentrations. Mechanistic categories:
(E) Conformational selection. The fuzzy N-terminal acidic tail of
the Max transcription factor (1nkp) facilitates formation of the DNA binding
helix (dark red) of the leucine zipper basic helix–loop–helix
(bHLH) motif. (F) Flexibility modulation. The disordered serine/arginine-rich
region of the Ets-1 transcription factor (1mdm) changes DNA binding affinity by 100–1000-fold
by modulating the flexibility of the binding segment via transient
interactions. (G) Competitive binding. The acidic fuzzy C-terminal
tail of high-mobility group protein B1 (2gzk) competes with DNA for the positively
charged binding surfaces. (H) Tethering. The binding of the virion
protein 16 activation domain to the human transcriptional coactivator
positive cofactor 4 (2phe) is facilitated by acidic disordered regions,
which anchor the binding segments.
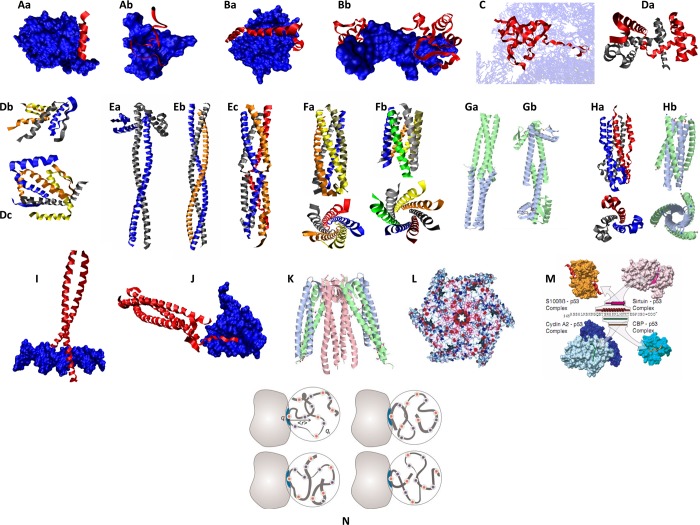
A portrait gallery of disorder-based complexes. Illustrative
examples
of various interaction modes of intrinsically disordered proteins
are shown. Protein Data Bank identifiers
for the structures are given in parentheses. (A) MoRFs. Aa, α-MoRF,
a complex between the botulinum neurotoxin (red helix) and its receptor
(a blue cloud) (2NM1); Ab, ι-MoRF, a complex between an 18-mer cognate peptide
derived from the α1 subunit of the nicotinic acetylcholine receptor
from Torpedo californica (red helix)
and α-cobratoxin (a blue cloud) (1LXH). (B) Wrappers. Ba, rat PP1 (blue cloud)
complexed with mouse inhibitor-2 (red helices) (2O8A); Bb, a complex
between the paired domain from the Drosophila paired (prd) protein and DNA (1PDN). (C) Penetrator. Ribosomal protein s12
embedded into the rRNA (1N34). (D) Huggers. Da, E. colitrp repressor dimer (1ZT9); Db, tetramerization domain of p53 (1PES); Dc, tetramerization
domain of p73 (2WQI). (E) Intertwined strings. Ea, dimeric coiled coil, a basic coiled-coil
protein from Eubacterium eligens ATCC
27750 (3HNW);
Eb, trimeric coiled coil, salmonella trimeric autotransporter adhesin,
SadA (2WPQ);
Ec, tetrameric coiled coil, the virion-associated protein P3 from
Caulimovirus (2O1J). (F) Long cylindrical containers. Fa, pentameric coiled coil, side
and top views of the assembly domain of cartilage oligomeric matrix
protein (1FBM); Fb, side and top views of the seven-helix coiled coil, engineered
version of the GCN4 leucine zipper (2HY6). (G) Connectors. Ga, human heat shock
factor binding protein 1 (3CI9); Gb, the bacterial cell division protein ZapA from Pseudomonas aeruginosa (1W2E). (H) Armature. Ha, side and top views
of the envelope glycoprotein GP2 from Ebola virus (2EBO); Hb, side and top
views of a complex between the N- and C-terminal peptides derived
from the membrane fusion protein of the Visna (1JEK). (I) Tweezers or
forceps. A complex between c-Jun, c-Fos, and DNA. Proteins are shown
as red helices, whereas DNA is shown as a blue cloud (1FOS). (J) Grabbers.
Structure of the complex between βPIX coiled coil (red helices)
and Shank PDZ (blue cloud) (3L4F). (K) Tentacles. Structure of the hexameric molecular
chaperone prefoldin from the archaeum Methanobacterium
thermoautotrophicum (1FXK). (L) Pullers. Structure of the ClpB
chaperone from Thermus thermophilus (1QVR). (M)
Chameleons. The C-terminal fragment of p53 gains different types of
secondary structure in complexes with four different binding partners,
cyclin A (1H26), sirtuin (1MA3), CBP bromo domain (1JSP), and s100ββ (1DT7). Panels A–M reprinted with permission
from ref (257). Copyright
2011 The Royal Society of Chemistry. (N) Dynamic complexes. Schematic
representation of the polyelectrostatic model of the Sic1–Cdc4
interaction. An IDP (ribbon) interacts with a folded receptor (gray
shape) through several distinct binding motifs and an ensemble of
conformations (indicated by four representations of the interaction).
The intrinsically disordered protein possesses positive and negative
charges (depicted as blue and red circles, respectively) giving rise
to a net charge ql, while the binding
site in the receptor (light blue) has a charge qr. The effective distance ⟨r⟩
is between the binding site and the center of mass of the intrinsically
disordered protein. Panel N was reprinted with permission from ref (243). Copyright 2010 John
Wiley & Sons, Inc.
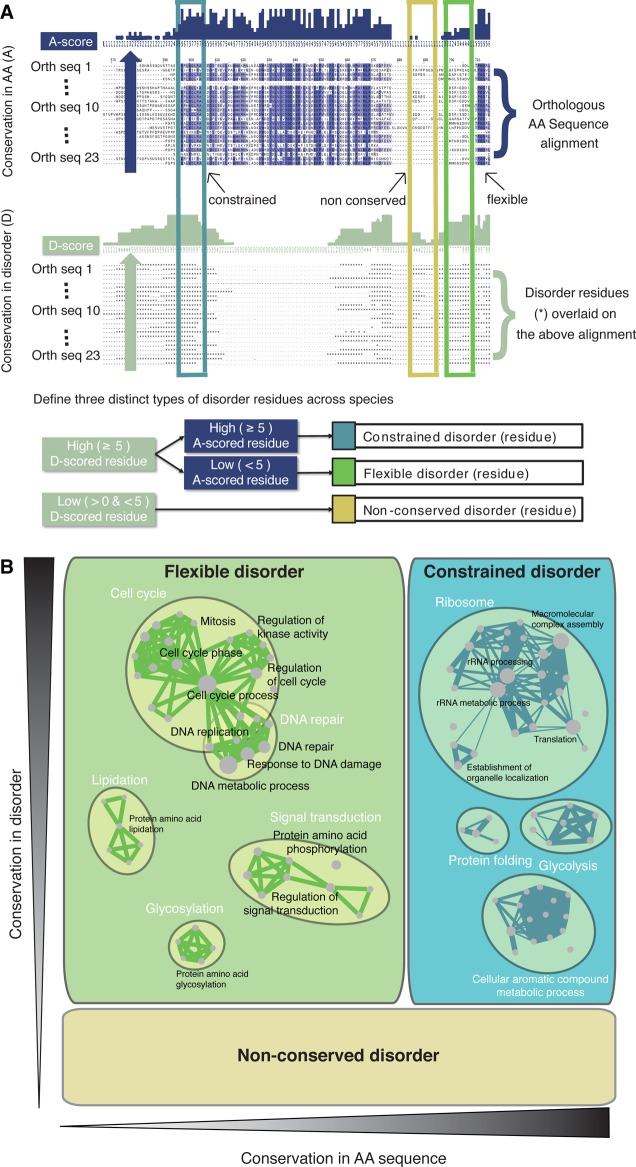
Classification
of disordered regions according to their evolutionary
conservation (constrained, flexible, and nonconserved disorder). (A)
Schematic of computing disorder conservation and amino acid sequence
conservation. The alignments are used to calculate the percentage
of sequences in which a residue is disordered and the percentage of
sequences in which the amino acid itself is conserved. A residue is
considered to be conserved disordered if the property of disorder
is conserved in at least one-half of the species. Similarly, the amino
acid type of a residue is considered conserved if it is present in
at least one-half of the species. Disordered residues in which both
sequence and disorder are conserved are referred to as constrained
disorder. Disordered residues in which disorder is conserved but not
the amino acid sequence are referred to as flexible disorder. Residues
that are disordered in S. cerevisiae but not cases of conserved disorder are referred to as nonconserved
disorder. (B) Disorder splits into three distinct phenomena. Functional
enrichment maps of proteins enriched in flexible disorder versus constrained
disorder. The area of each rectangle is proportional to the occurrence
of that type of disorder in the alignments. Related gene ontology
terms are grouped based on gene overlap. Reprinted with permission
from ref (54). Copyright
2011 Springer Science + Business Media.
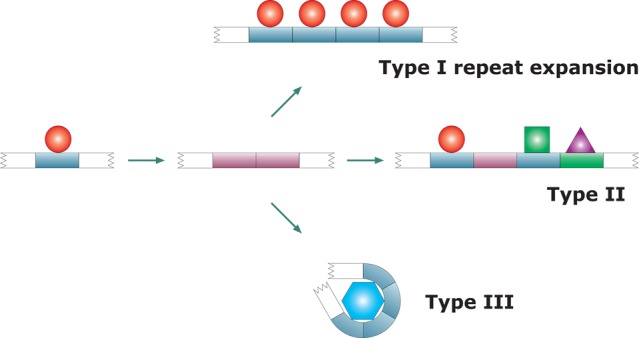
Repeat expansion
creates IDRs. IDRs are abundant in repeating sequence
elements, which suggests that repeat expansion is an important mechanism
by which genetic material encoding for structural disorder is generated.
The expanding repeats may fall into three classes (types) in terms
of their functional diversification following expansion. Individual
repeats may remain functionally equivalent (type I), or diversify
(type II), or collectively acquire a completely new function (type
III). Dark-tone red indicates structural disorder of the repeat, which
may undergo full (dark-tone blue) or partial (green) induced folding
upon binding to a partner. Adapted with permission from ref (61). Copyright 2003 John Wiley
& Sons, Inc.
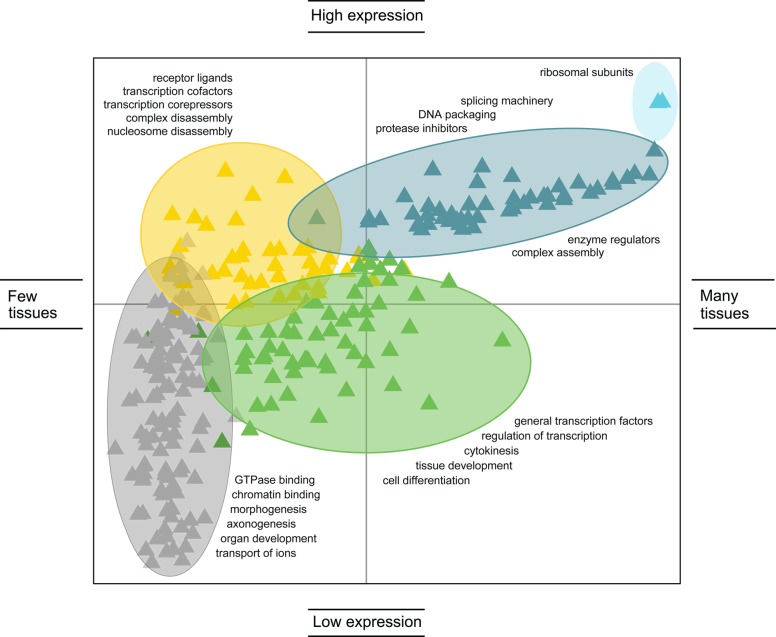
A summary of expression–function trends for human
transcripts
encoding highly disordered proteins. The x-axis represents
the log10 number of tissues in which the transcript is
expressed; the y-axis represents the log10 average magnitude of expression within the tissues. From the data,
five distinct functional classes of highly disordered human proteins
become apparent. Adapted with permission from ref (208). Copyright 2009 Springer
Science + Business Media.
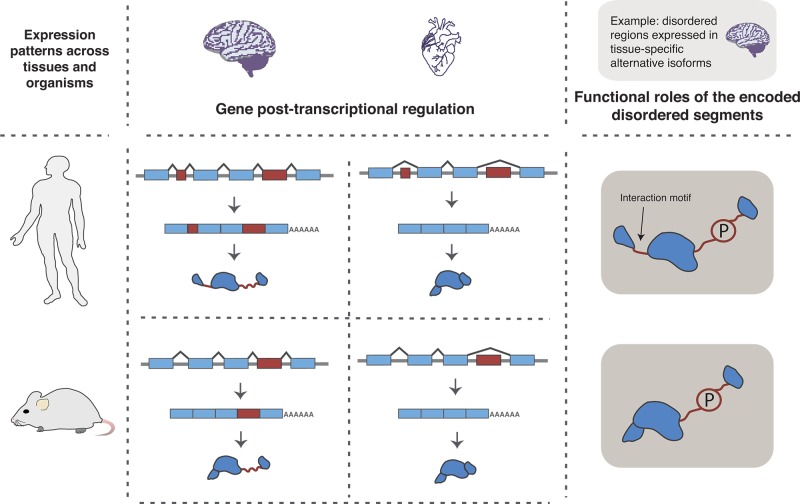
Transcriptional and post-transcriptional gene
regulation can be
informative of IDR function. How inclusion of exons that code for
IDRs is regulated during gene transcription and alternative splicing
can give insights into the functional roles of the encoded disordered
regions. For example, tissue- or developmental-specific regulation
of alternative splicing or alternative promoter and polyadenylation
site usage can be associated with important roles of the encoded IDRs
in protein regulation and cellular interactions through, for example,
the presence of binding motifs and phosphosites. Additionally, information
on the conservation of patterns of exon inclusion (i.e., events shared
among different evolutionary lineages versus species-specific events)
can aid in better characterization of the encoded IDRs. The figure
illustrates a hypothetical example where an exon (largest red box)
that is included in a tissue-specific manner both in human and in
mouse encodes an IDR that embeds a phosphosite (P) and is involved
in protein regulation. The human gene depicted in the figure has an
additional exon (smallest red box), which encodes an IDR with a short
interaction motif and which is also included in a tissue-specific
manner in humans. Gene structures, mature mRNAs, and corresponding
protein isoforms are shown for human and mouse brain and heart tissues.
On the right, possible functional roles of the IDRs encoded by the
brain isoforms are illustrated. The examples illustrate how protein
functional space can increase due to alternative splicing of exons
that encode IDRs. Adapted with permission from ref (304). Copyright 2012 Elsevier.
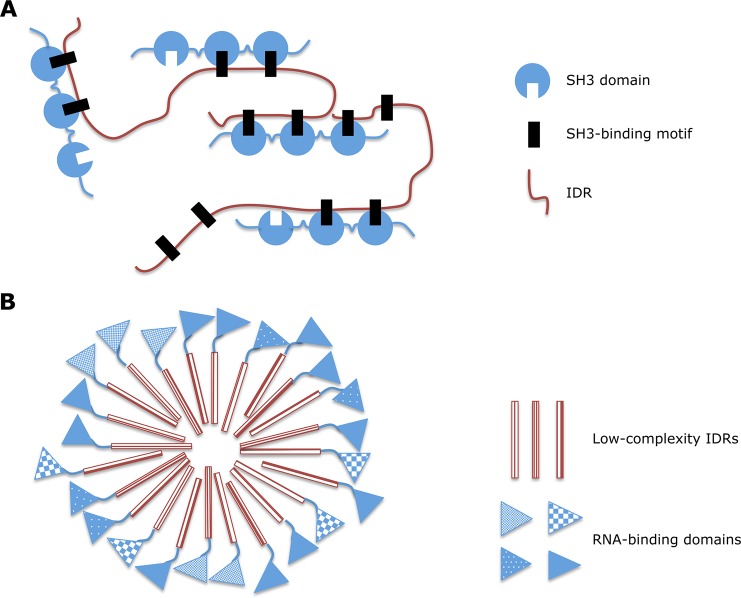
Involvement of IDRs
in phase transitions. (A) Interactions between
proteins that contain multiple copies of a specific domain (an SH3
domain in the figure) and IDRs with multiple instances of its interaction
motif (proline-rich SH3 motif here) can, at appropriate concentrations,
produce sharp liquid–liquid-demixing phase separations. This
phase transition is likely to increase local “active”
protein concentrations exploitable for signaling switches. (B) High
concentrations of low-complexity IDRs found in certain RNA binding
domains lead to a reversible phase transition with the formation of
highly dynamic hydrogels. These RNA granule-like assemblies consist
of heteromeric protein aggregates and allow localization and storage
of functionally related but nonidentical RNA molecules. Adapted from
ref (100). Copyright
2013 the Biochemical Society.
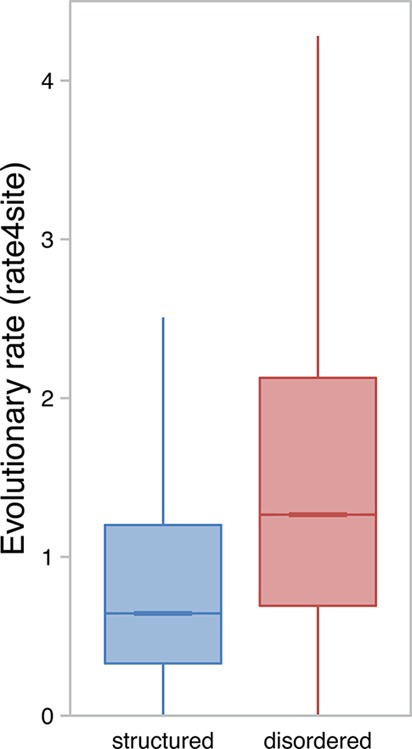
Boxplots of the
distributions of evolutionary rates for predicted
structured (blue) and disordered (red) residues across the human proteome.
Residues with a high evolutionary rate are less conserved. Boxes represent
the 50% of data points in the two quartiles above and below the median
(the horizontal bar within each box). Vertical lines (whiskers) connected
to the boxes represent the highest and lowest nonoutlier data points,
with outliers being defined as >1.5 times the interquartile range
from the median. Outliers are not shown for visual clarity.
Similar articles
-
Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs).Chem Rev. 2014 Jul 9;114(13):6661-714. doi: 10.1021/cr400695p. Epub 2014 Jun 5. Chem Rev. 2014. PMID: 24901537 Free PMC article. Review. No abstract available.
-
Functional roles of transiently and intrinsically disordered regions within proteins.FEBS J. 2015 Apr;282(7):1182-9. doi: 10.1111/febs.13202. Epub 2015 Jan 29. FEBS J. 2015. PMID: 25631540 Review.
-
Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function.Chem Rev. 2014 Jul 9;114(13):6779-805. doi: 10.1021/cr400459c. Epub 2014 Feb 6. Chem Rev. 2014. PMID: 24502763 Free PMC article. Review. No abstract available.
-
Sequence and Structure Properties Uncover the Natural Classification of Protein Complexes Formed by Intrinsically Disordered Proteins via Mutual Synergistic Folding.Int J Mol Sci. 2019 Nov 1;20(21):5460. doi: 10.3390/ijms20215460. Int J Mol Sci. 2019. PMID: 31683980 Free PMC article.
-
A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction.Brief Bioinform. 2019 Jan 18;20(1):330-346. doi: 10.1093/bib/bbx126. Brief Bioinform. 2019. PMID: 30657889 Review.
Cited by
-
Analysis of Protein Disorder Predictions in the Light of a Protein Structural Alphabet.Biomolecules. 2020 Jul 20;10(7):1080. doi: 10.3390/biom10071080. Biomolecules. 2020. PMID: 32698546 Free PMC article.
-
SENSE-PPI reconstructs interactomes within, across, and between species at the genome scale.iScience. 2024 Jun 25;27(7):110371. doi: 10.1016/j.isci.2024.110371. eCollection 2024 Jul 19. iScience. 2024. PMID: 39055916 Free PMC article.
-
Nuclear bodies: the emerging biophysics of nucleoplasmic phases.Curr Opin Cell Biol. 2015 Jun;34:23-30. doi: 10.1016/j.ceb.2015.04.003. Epub 2015 May 15. Curr Opin Cell Biol. 2015. PMID: 25942753 Free PMC article. Review.
-
Protein conformation and biomolecular condensates.Curr Res Struct Biol. 2022 Sep 14;4:285-307. doi: 10.1016/j.crstbi.2022.09.004. eCollection 2022. Curr Res Struct Biol. 2022. PMID: 36164646 Free PMC article. Review.
-
The HSV-1 Transcription Factor ICP4 Confers Liquid-Like Properties to Viral Replication Compartments.Int J Mol Sci. 2021 Apr 24;22(9):4447. doi: 10.3390/ijms22094447. Int J Mol Sci. 2021. PMID: 33923223 Free PMC article.
References
-
- Flicek P.; Ahmed I.; Amode M. R.; Barrell D.; Beal K.; Brent S.; Carvalho-Silva D.; Clapham P.; Coates G.; Fairley S.; Fitzgerald S.; Gil L.; Garcia-Giron C.; Gordon L.; Hourlier T.; Hunt S.; Juettemann T.; Kahari A. K.; Keenan S.; Komorowska M.; Kulesha E.; Longden I.; Maurel T.; McLaren W. M.; Muffato M.; Nag R.; Overduin B.; Pignatelli M.; Pritchard B.; Pritchard E.; Riat H. S.; Ritchie G. R.; Ruffier M.; Schuster M.; Sheppard D.; Sobral D.; Taylor K.; Thormann A.; Trevanion S.; White S.; Wilder S. P.; Aken B. L.; Birney E.; Cunningham F.; Dunham I.; Harrow J.; Herrero J.; Hubbard T. J.; Johnson N.; Kinsella R.; Parker A.; Spudich G.; Yates A.; Zadissa A.; Searle S. M. Nucleic Acids Res. 2013, 41, D48. - PMC - PubMed
-
- Kolodny R.; Pereyaslavets L.; Samson A. O.; Levitt M. Annu. Rev. Biophys. 2013, 42, 559. - PubMed
-
- Raes J.; Harrington E. D.; Singh A. H.; Bork P. Curr. Opin. Struct. Biol. 2007, 17, 362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
