

Pigment epithelium-derived factor 34-mer peptide prevents liver fibrosis and hepatic stellate cell activation through down-regulation of the PDGF receptor
- PMID: 24763086
- PMCID: PMC3998957
- DOI: 10.1371/journal.pone.0095443
Pigment epithelium-derived factor 34-mer peptide prevents liver fibrosis and hepatic stellate cell activation through down-regulation of the PDGF receptor
Erratum in
- PLoS One. 2014;9(9): doi/10.1371/journal.pone.0108835
Abstract
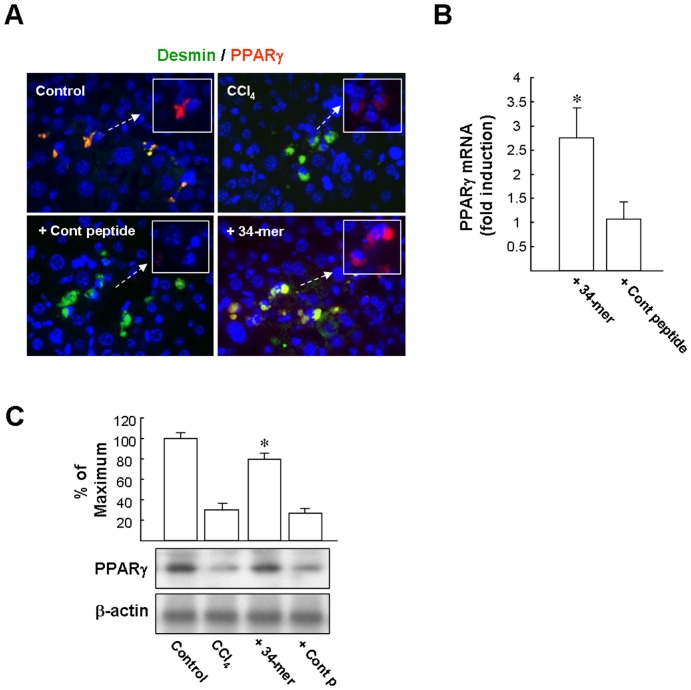
Pigment epithelium-derived factor (PEDF) has been shown previously to prevent liver fibrosis and hepatic stellate cell (HSC) activation. By investigating the functional domains in PEDF, we identified a 34-mer peptide (residues Asp44-Asn77) that harbors the same function as the full-length PEDF protein. Not only did the 34-mer suppress the development of fibrosis in carbon tetrachloride (CCl4)-treated mouse liver but it also upregulated peroxisome proliferator-activated receptor-gamma (PPARγ) expression in HSCs in vivo. Platelet-derived growth factor (PDGF) plays a crucial role on the process of HSC activation in response to liver damage. The 34-mer suppressed PDGF-induced cell proliferation and expression of myofibroblastic marker proteins in primary rat HSC culture, increased the levels of PPARγ mRNA and protein in a dose-dependent manner and markedly reduced the level of active β-catenin protein, an HSC activating factor, in HSC-T6 cells. Similarly, IWR-1, an inhibitor of the Wnt response, displayed the same effect as the 34-mer in preventing HSC-T6 activation. The Wnt signaling-mediated PPARγ suppression was abolished by both the IWR-1 inhibitor and a small interfering RNA (siRNA) targeting β-catenin and the Wnt coreceptor, LRP6. Both PEDF and the 34-mer down-regulated PDGF receptor-α/β expression and blocked the PDGF-induced phosphorylation of Akt and ERK. Moreover, the inhibitory effect on PDGF receptor expression was abolished by PPARγ antagonists and PPARγ siRNA. Our observations indicate that the PEDF-derived 34-mer peptide can mimic PEDF in attenuating HSC activation. Investigation of this 34-mer peptide led to the identification of a signaling mechanism involving PPARγ induction, suppression of Wnt/β-catenin signaling and down-regulation of the PDGF receptor-α/β.
Conflict of interest statement
Figures






Similar articles
-
Pigment epithelium-derived factor is an intrinsic antifibrosis factor targeting hepatic stellate cells.Am J Pathol. 2010 Oct;177(4):1798-811. doi: 10.2353/ajpath.2010.091085. Epub 2010 Aug 13. Am J Pathol. 2010. PMID: 20709803 Free PMC article.
-
Pigment Epithelium Derived Factor Peptide Protects Murine Hepatocytes from Carbon Tetrachloride-Induced Injury.PLoS One. 2016 Jul 6;11(7):e0157647. doi: 10.1371/journal.pone.0157647. eCollection 2016. PLoS One. 2016. PMID: 27384427 Free PMC article.
-
Selective inhibition of activated stellate cells and protection from carbon tetrachloride-induced liver injury in rats by a new PPARgamma agonist KR62776.Arch Pharm Res. 2010 Mar;33(3):433-42. doi: 10.1007/s12272-010-0313-3. Epub 2010 Mar 30. Arch Pharm Res. 2010. PMID: 20361309 Free PMC article.
-
Wnt signaling in liver fibrosis: progress, challenges and potential directions.Biochimie. 2013 Dec;95(12):2326-35. doi: 10.1016/j.biochi.2013.09.003. Epub 2013 Sep 13. Biochimie. 2013. PMID: 24036368 Review.
-
Peroxisome proliferator-activated receptor-γ as a therapeutic target for hepatic fibrosis: from bench to bedside.Cell Mol Life Sci. 2013 Jan;70(2):259-76. doi: 10.1007/s00018-012-1046-x. Epub 2012 Jun 15. Cell Mol Life Sci. 2013. PMID: 22699820 Free PMC article. Review.
Cited by
-
Pigment epithelium-derived factor (PEDF) normalizes matrix defects in iPSCs derived from Osteogenesis imperfecta Type VI.Rare Dis. 2016 Jul 19;4(1):e1212150. doi: 10.1080/21675511.2016.1212150. eCollection 2016. Rare Dis. 2016. PMID: 27579219 Free PMC article.
-
Pigment Epithelium-Derived Factor (PEDF) as a Regulator of Wound Angiogenesis.Sci Rep. 2018 Jul 24;8(1):11142. doi: 10.1038/s41598-018-29465-9. Sci Rep. 2018. PMID: 30042381 Free PMC article.
-
Melanoma Cells Block PEDF Production in Fibroblasts to Induce the Tumor-Promoting Phenotype of Cancer-Associated Fibroblasts.Cancer Res. 2016 Apr 15;76(8):2265-76. doi: 10.1158/0008-5472.CAN-15-2468. Epub 2016 Feb 26. Cancer Res. 2016. PMID: 26921338 Free PMC article.
-
Scleroderma fibroblasts suppress angiogenesis via TGF-β/caveolin-1 dependent secretion of pigment epithelium-derived factor.Ann Rheum Dis. 2018 Mar;77(3):431-440. doi: 10.1136/annrheumdis-2017-212120. Epub 2017 Dec 19. Ann Rheum Dis. 2018. PMID: 29259049 Free PMC article.
-
Pigment Epithelium-Derived Factor (PEDF) is a Determinant of Stem Cell Fate: Lessons from an Ultra-Rare Disease.J Dev Biol. 2015 Dec;3(4):112-128. doi: 10.3390/jdb3040112. Epub 2015 Nov 20. J Dev Biol. 2015. PMID: 27239449 Free PMC article.
References
-
- Friedman SL (2000) Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275(4): 2247–2250. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
