

Template-based structure modeling of protein-protein interactions
- PMID: 24721449
- PMCID: PMC3984454
- DOI: 10.1016/j.sbi.2013.11.005
Template-based structure modeling of protein-protein interactions
Abstract
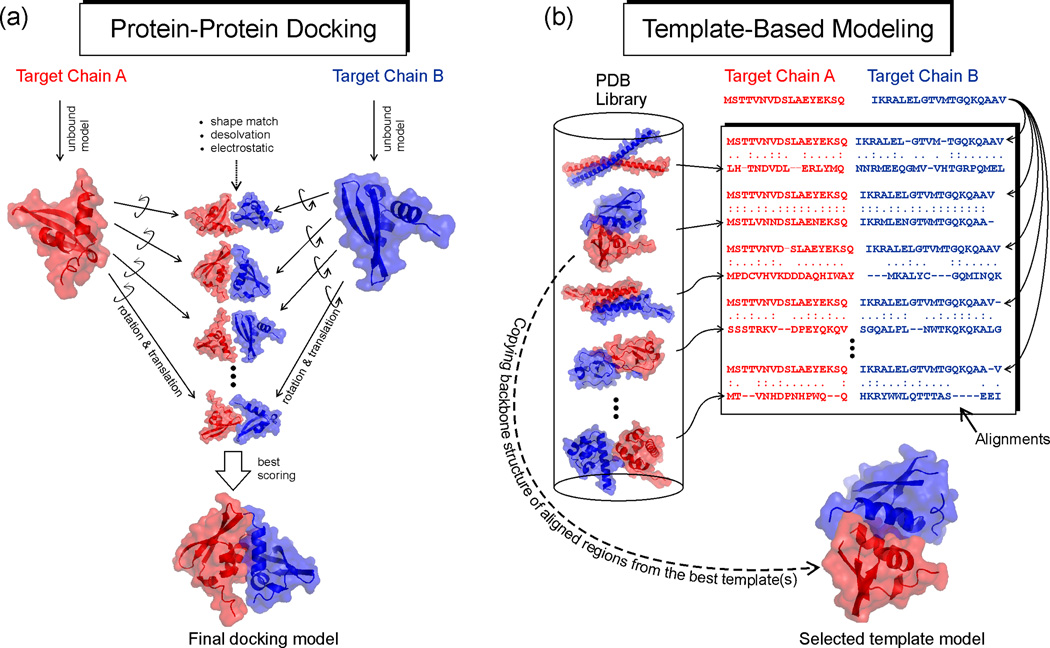
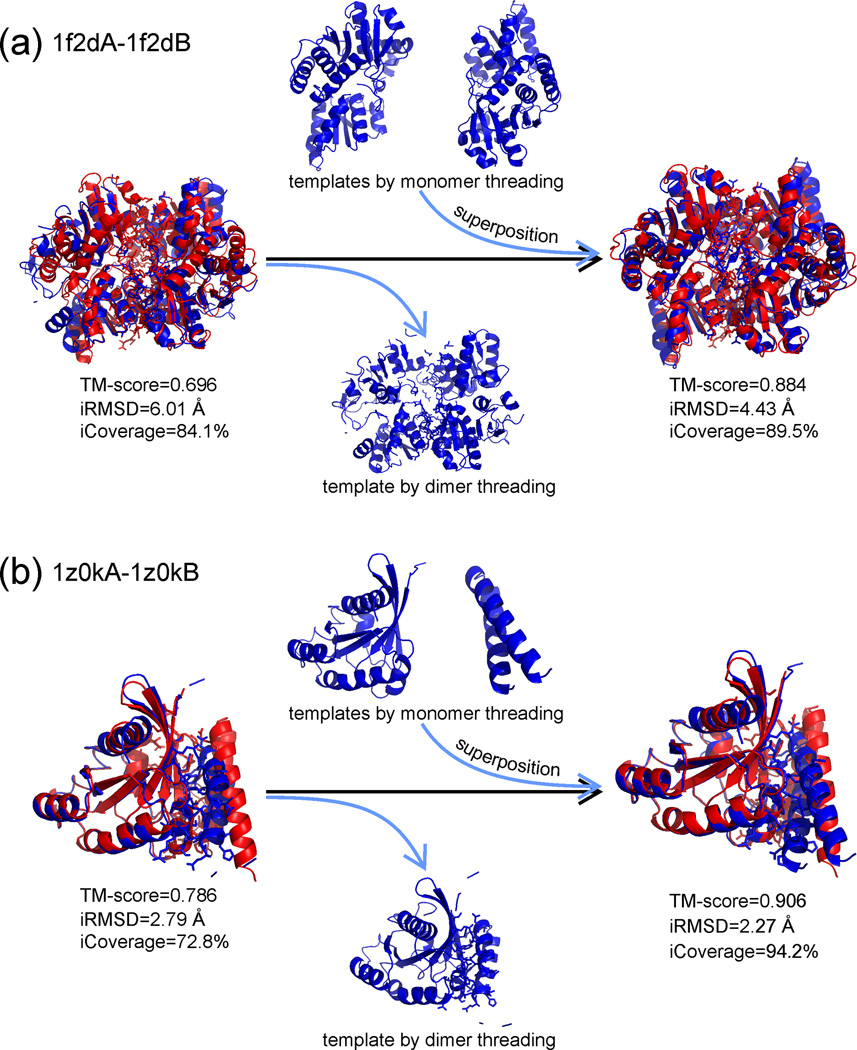
The structure of protein-protein complexes can be constructed by using the known structure of other protein complexes as a template. The complex structure templates are generally detected either by homology-based sequence alignments or, given the structure of monomer components, by structure-based comparisons. Critical improvements have been made in recent years by utilizing interface recognition and by recombining monomer and complex template libraries. Encouraging progress has also been witnessed in genome-wide applications of template-based modeling, with modeling accuracy comparable to high-throughput experimental data. Nevertheless, bottlenecks exist due to the incompleteness of the protein-protein complex structure library and the lack of methods for distant homologous template identification and full-length complex structure refinement.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures

Similar articles
-
Template-Based Modeling of Protein Complexes Using the PPI3D Web Server.Methods Mol Biol. 2020;2165:139-155. doi: 10.1007/978-1-0716-0708-4_8. Methods Mol Biol. 2020. PMID: 32621223
-
How to choose templates for modeling of protein complexes: Insights from benchmarking template-based docking.Proteins. 2020 Aug;88(8):1070-1081. doi: 10.1002/prot.25875. Epub 2020 Feb 7. Proteins. 2020. PMID: 31994759 Free PMC article.
-
Accuracy of protein-protein binding sites in high-throughput template-based modeling.PLoS Comput Biol. 2010 Apr 1;6(4):e1000727. doi: 10.1371/journal.pcbi.1000727. PLoS Comput Biol. 2010. PMID: 20369011 Free PMC article.
-
Advances in template-based protein docking by utilizing interfaces towards completing structural interactome.Curr Opin Struct Biol. 2015 Dec;35:87-92. doi: 10.1016/j.sbi.2015.10.001. Epub 2015 Nov 9. Curr Opin Struct Biol. 2015. PMID: 26539658 Review.
-
A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction.Curr Opin Struct Biol. 2005 Jun;15(3):285-9. doi: 10.1016/j.sbi.2005.05.011. Curr Opin Struct Biol. 2005. PMID: 15939584 Review.
Cited by
-
Struct2Graph: a graph attention network for structure based predictions of protein-protein interactions.BMC Bioinformatics. 2022 Sep 10;23(1):370. doi: 10.1186/s12859-022-04910-9. BMC Bioinformatics. 2022. PMID: 36088285 Free PMC article.
-
Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data.Nat Methods. 2024 Feb;21(2):279-289. doi: 10.1038/s41592-023-02130-4. Epub 2024 Jan 2. Nat Methods. 2024. PMID: 38167654 Free PMC article.
-
Multiple kernels learning-based biological entity relationship extraction method.J Biomed Semantics. 2017 Sep 20;8(Suppl 1):38. doi: 10.1186/s13326-017-0138-9. J Biomed Semantics. 2017. PMID: 29297359 Free PMC article.
-
Structural templates for comparative protein docking.Proteins. 2015 Sep;83(9):1563-70. doi: 10.1002/prot.24736. Epub 2015 Jun 13. Proteins. 2015. PMID: 25488330 Free PMC article.
-
Protein-protein interaction prediction with deep learning: A comprehensive review.Comput Struct Biotechnol J. 2022 Sep 19;20:5316-5341. doi: 10.1016/j.csbj.2022.08.070. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36212542 Free PMC article. Review.
References
-
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. - PubMed
-
- Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. - PubMed
-
- Mosca R, Pons T, Ceol A, Valencia A, Aloy P. Towards a detailed atlas of protein-protein interactions. Curr Opin Struct Biol. 2013 - PubMed
-
- Stein A, Mosca R, Aloy P. Three-dimensional modeling of protein interactions and complexes is going 'omics. Curr Opin Struct Biol. 2011;21:200–208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
