

Evolutionary genomics of Borrelia burgdorferi sensu lato: findings, hypotheses, and the rise of hybrids
- PMID: 24704760
- PMCID: PMC4299872
- DOI: 10.1016/j.meegid.2014.03.025
Evolutionary genomics of Borrelia burgdorferi sensu lato: findings, hypotheses, and the rise of hybrids
Abstract
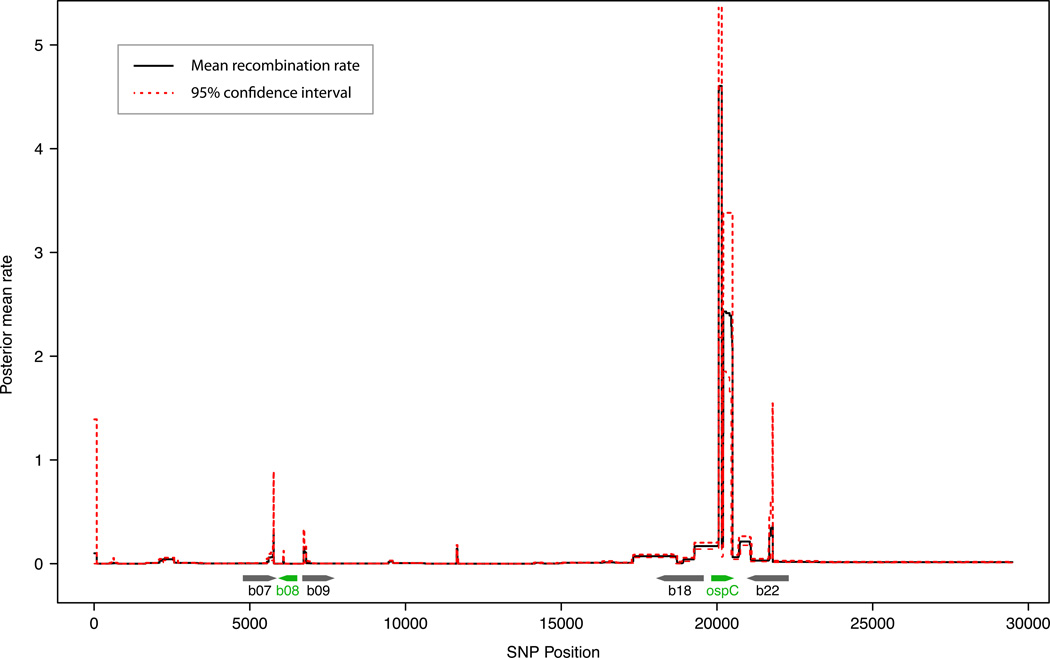
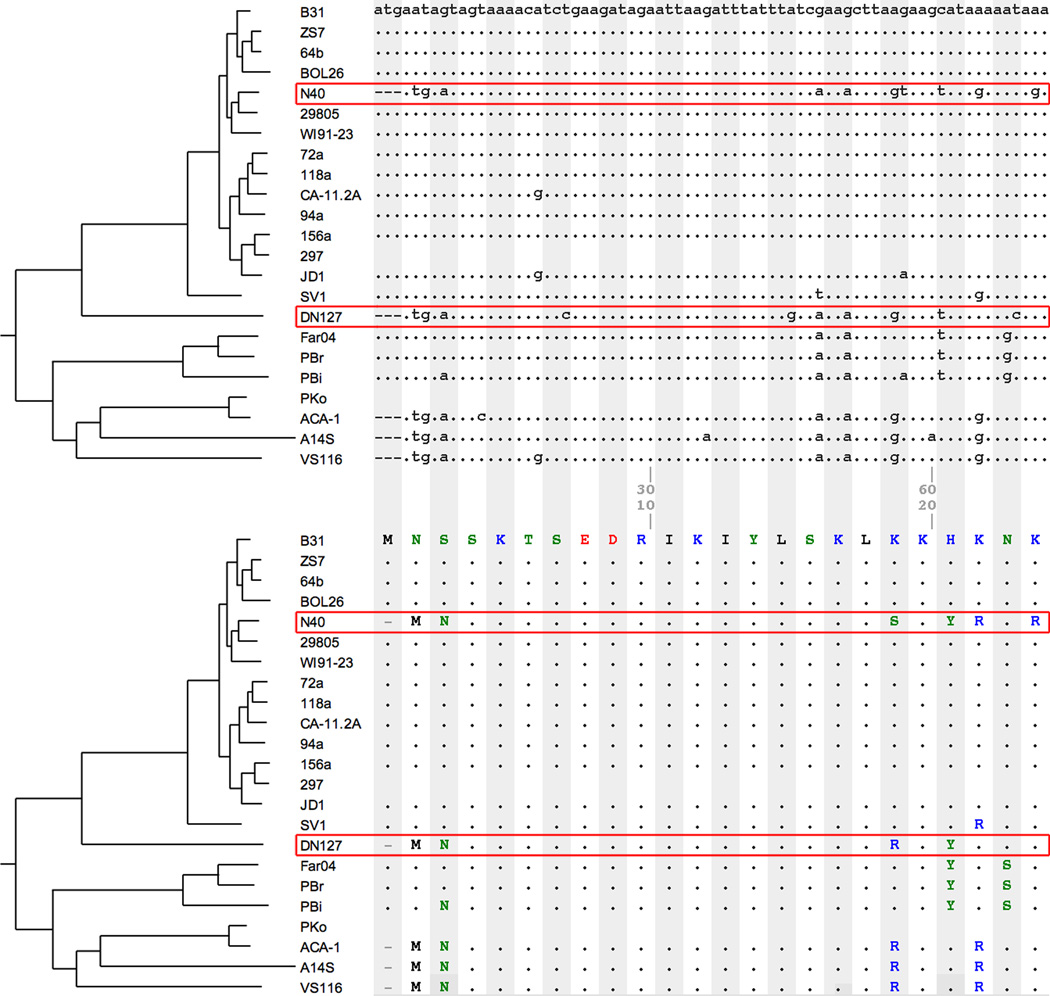
Borrelia burgdorferi sensu lato (B. burgdorferi s.l.), the group of bacterial species represented by Lyme disease pathogens, has one of the most complex and variable genomic architectures among prokaryotes. Showing frequent recombination within and limited gene flow among geographic populations, the B. burgdorferi s.l. genomes provide an excellent window into the processes of bacterial evolution at both within- and between-population levels. Comparative analyses of B. burgdorferi s.l. genomes revealed a highly dynamic plasmid composition but a conservative gene repertoire. Gene duplication and loss as well as sequence variations at loci encoding surface-localized lipoproteins (e.g., the PF54 genes) are strongly associated with adaptive differences between species. There are a great many conserved intergenic spacer sequences that are candidates for cis-regulatory elements and non-coding RNAs. Recombination among coexisting strains occurs at a rate approximately three times the mutation rate. The coexistence of a large number of genomic groups within local B. burgdorferi s.l. populations may be driven by immune-mediated diversifying selection targeting major antigen loci as well as by adaptation to multiple host species. Questions remain regarding the ecological causes (e.g., climate change, host movements, or new adaptations) of the ongoing range expansion of B. burgdorferi s.l. and on the genomic variations associated with its ecological and clinical variability. Anticipating an explosive growth of the number of B. burgdorferi s.l. genomes sampled from both within and among species, we propose genome-based methods to test adaptive mechanisms and to identify molecular bases of phenotypic variations. Genome sequencing is also necessary for monitoring a likely increase of genetic admixture of previously isolated species and populations in North America and elsewhere.
Keywords: Bacteria evolution; Lyme disease; Phylogenomics; Population genomics; Recombination.
Copyright © 2014. Published by Elsevier B.V.
Figures

Similar articles
-
Evolution and population genomics of the Lyme borreliosis pathogen, Borrelia burgdorferi.Trends Genet. 2015 Apr;31(4):201-7. doi: 10.1016/j.tig.2015.02.006. Epub 2015 Mar 9. Trends Genet. 2015. PMID: 25765920 Free PMC article. Review.
-
Natural selection and recombination at host-interacting lipoprotein loci drive genome diversification of Lyme disease and related bacteria.mBio. 2024 Sep 11;15(9):e0174924. doi: 10.1128/mbio.01749-24. Epub 2024 Aug 15. mBio. 2024. PMID: 39145656 Free PMC article.
-
Pervasive recombination and sympatric genome diversification driven by frequency-dependent selection in Borrelia burgdorferi, the Lyme disease bacterium.Genetics. 2011 Nov;189(3):951-66. doi: 10.1534/genetics.111.130773. Epub 2011 Sep 2. Genetics. 2011. PMID: 21890743 Free PMC article.
-
Inter- and intra-specific pan-genomes of Borrelia burgdorferi sensu lato: genome stability and adaptive radiation.BMC Genomics. 2013 Oct 10;14:693. doi: 10.1186/1471-2164-14-693. BMC Genomics. 2013. PMID: 24112474 Free PMC article.
-
Updates on Borrelia burgdorferi sensu lato complex with respect to public health.Ticks Tick Borne Dis. 2011 Sep;2(3):123-8. doi: 10.1016/j.ttbdis.2011.04.002. Epub 2011 May 27. Ticks Tick Borne Dis. 2011. PMID: 21890064 Free PMC article. Review.
Cited by
-
Phylogenomic identification of regulatory sequences in bacteria: an analysis of statistical power and an application to Borrelia burgdorferi sensu lato.mBio. 2015 Apr 14;6(2):e00011-15. doi: 10.1128/mBio.00011-15. mBio. 2015. PMID: 25873371 Free PMC article.
-
Infection history of the blood-meal host dictates pathogenic potential of the Lyme disease spirochete within the feeding tick vector.PLoS Pathog. 2018 Apr 5;14(4):e1006959. doi: 10.1371/journal.ppat.1006959. eCollection 2018 Apr. PLoS Pathog. 2018. PMID: 29621350 Free PMC article.
-
Evolution and population genomics of the Lyme borreliosis pathogen, Borrelia burgdorferi.Trends Genet. 2015 Apr;31(4):201-7. doi: 10.1016/j.tig.2015.02.006. Epub 2015 Mar 9. Trends Genet. 2015. PMID: 25765920 Free PMC article. Review.
-
Natural selection and recombination at host-interacting lipoprotein loci drive genome diversification of Lyme disease and related bacteria.mBio. 2024 Sep 11;15(9):e0174924. doi: 10.1128/mbio.01749-24. Epub 2024 Aug 15. mBio. 2024. PMID: 39145656 Free PMC article.
-
Primordial origin and diversification of plasmids in Lyme disease agent bacteria.BMC Genomics. 2018 Mar 27;19(1):218. doi: 10.1186/s12864-018-4597-x. BMC Genomics. 2018. PMID: 29580205 Free PMC article.
References
-
- Avise JC. Phylogeography: The History and Formation of Species. Harvard University Press; 2000.
-
- Barton NH. Why sex and recombination? Cold Spring Harb. Symp. Quant. Biol. 2009;74:187–195. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
