

Small-molecule inhibitors of the Myc oncoprotein
- PMID: 24657798
- PMCID: PMC4169356
- DOI: 10.1016/j.bbagrm.2014.03.005
Small-molecule inhibitors of the Myc oncoprotein
Abstract
The c-Myc (Myc) oncoprotein is among the most attractive of cancer targets given that it is de-regulated in the majority of tumors and that its inhibition profoundly affects their growth and/or survival. However, its role as a seldom-mutated transcription factor, its lack of enzymatic activity for which suitable pharmaceutical inhibitors could be crafted and its expression by normal cells have largely been responsible for its being viewed as "undruggable". Work over the past several years, however, has begun to reverse this idea by allowing us to view Myc within the larger context of global gene regulatory control. Thus, Myc and its obligate heterodimeric partner, Max, are integral to the coordinated recruitment and post-translational modification of components of the core transcriptional machinery. Moreover, Myc over-expression re-programs numerous critical cellular functions and alters the cell's susceptibility to their inhibition. This new knowledge has therefore served as a framework upon which to develop new pharmaceutical approaches. These include the continuing development of small molecules which act directly to inhibit the critical Myc-Max interaction, those which act indirectly to prevent Myc-directed post-translational modifications necessary to initiate productive transcription and those which inhibit vital pathways upon which the Myc-transformed cell is particularly reliant. This article is part of a Special Issue entitled: Myc proteins in cell biology and pathology.
Keywords: Cancer; Max; Myc; Protein–protein interaction; Small-molecule inhibitor; Transcription factor.
Copyright © 2014 Elsevier B.V. All rights reserved.
Figures
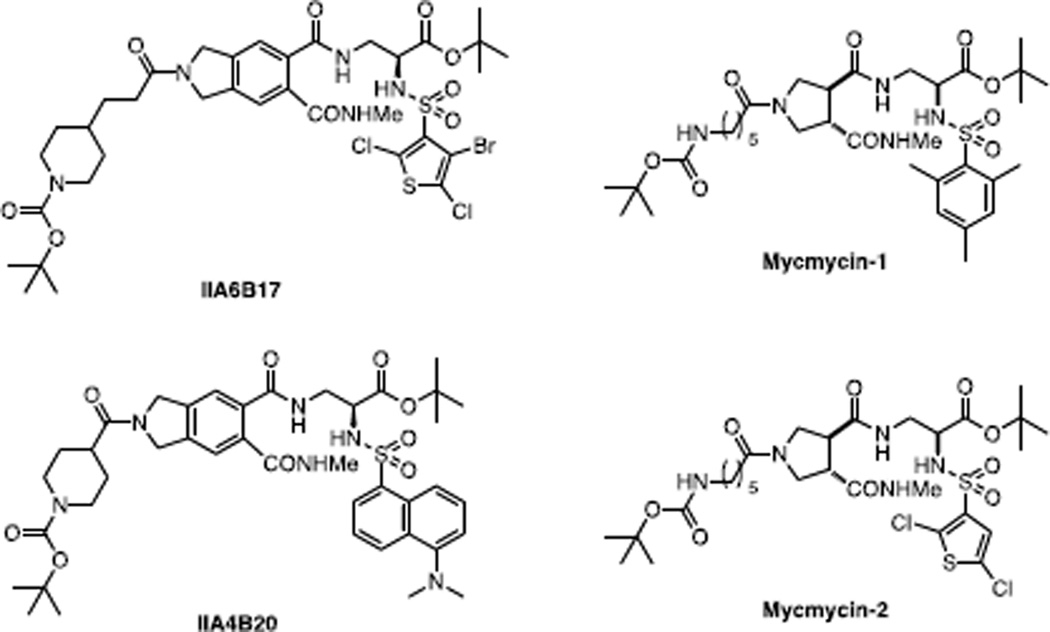
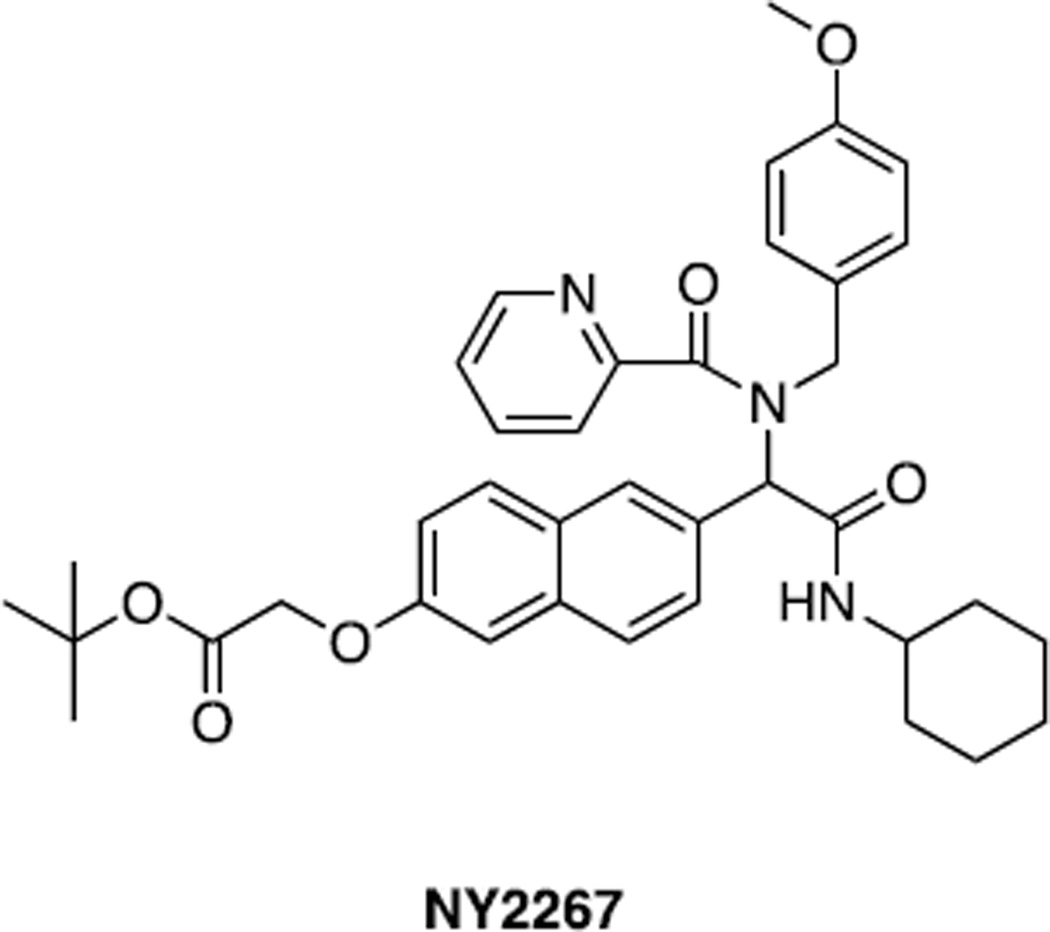
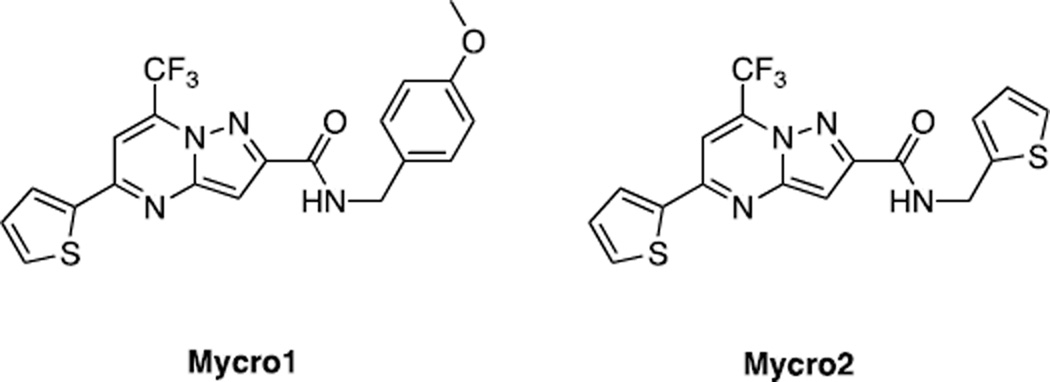
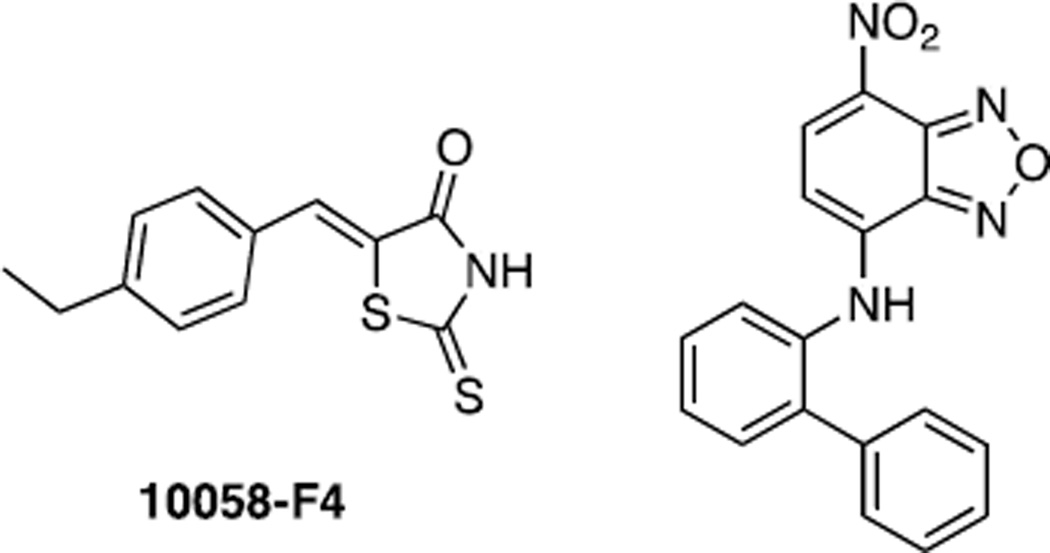
Similar articles
-
The activities of MYC, MNT and the MAX-interactome in lymphocyte proliferation and oncogenesis.Biochim Biophys Acta. 2015 May;1849(5):554-62. doi: 10.1016/j.bbagrm.2014.04.004. Epub 2014 Apr 13. Biochim Biophys Acta. 2015. PMID: 24731854 Review.
-
Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis.Biochim Biophys Acta. 2015 May;1849(5):484-500. doi: 10.1016/j.bbagrm.2014.05.016. Epub 2014 May 22. Biochim Biophys Acta. 2015. PMID: 24857747 Free PMC article. Review.
-
In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization.J Pharmacol Exp Ther. 2010 Dec;335(3):715-27. doi: 10.1124/jpet.110.170555. Epub 2010 Aug 26. J Pharmacol Exp Ther. 2010. PMID: 20801893 Free PMC article.
-
Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids.Oncotarget. 2015 Oct 20;6(32):32380-95. doi: 10.18632/oncotarget.6116. Oncotarget. 2015. PMID: 26474287 Free PMC article.
-
Myc and its interactors take shape.Biochim Biophys Acta. 2015 May;1849(5):469-83. doi: 10.1016/j.bbagrm.2014.06.002. Epub 2014 Jun 14. Biochim Biophys Acta. 2015. PMID: 24933113 Review.
Cited by
-
Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion.Oncotarget. 2015 Jun 30;6(18):15857-70. doi: 10.18632/oncotarget.4327. Oncotarget. 2015. PMID: 26036281 Free PMC article.
-
Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy.Cancer Cell. 2019 Nov 11;36(5):483-497.e15. doi: 10.1016/j.ccell.2019.10.001. Epub 2019 Oct 31. Cancer Cell. 2019. PMID: 31679823 Free PMC article.
-
Normal and Neoplastic Growth Suppression by the Extended Myc Network.Cells. 2022 Feb 21;11(4):747. doi: 10.3390/cells11040747. Cells. 2022. PMID: 35203395 Free PMC article. Review.
-
Functional inhibition of c-Myc using novel inhibitors identified through "hot spot" targeting.J Biol Chem. 2022 May;298(5):101898. doi: 10.1016/j.jbc.2022.101898. Epub 2022 Apr 1. J Biol Chem. 2022. PMID: 35378126 Free PMC article.
-
Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy.Sci Transl Med. 2019 Mar 20;11(484):eaar5012. doi: 10.1126/scitranslmed.aar5012. Sci Transl Med. 2019. PMID: 30894502 Free PMC article.
References
-
- Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin. Cancer Biol. 2006;16:313–317. - PubMed
-
- Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. - PubMed
-
- Pelengaris S, Khan M. The many faces of c-MYC. Arch. Biochem. Biophys. 2003;416:129–136. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
