

Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection
- PMID: 24646941
- PMCID: PMC3960176
- DOI: 10.1371/journal.pone.0092140
Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection
Abstract
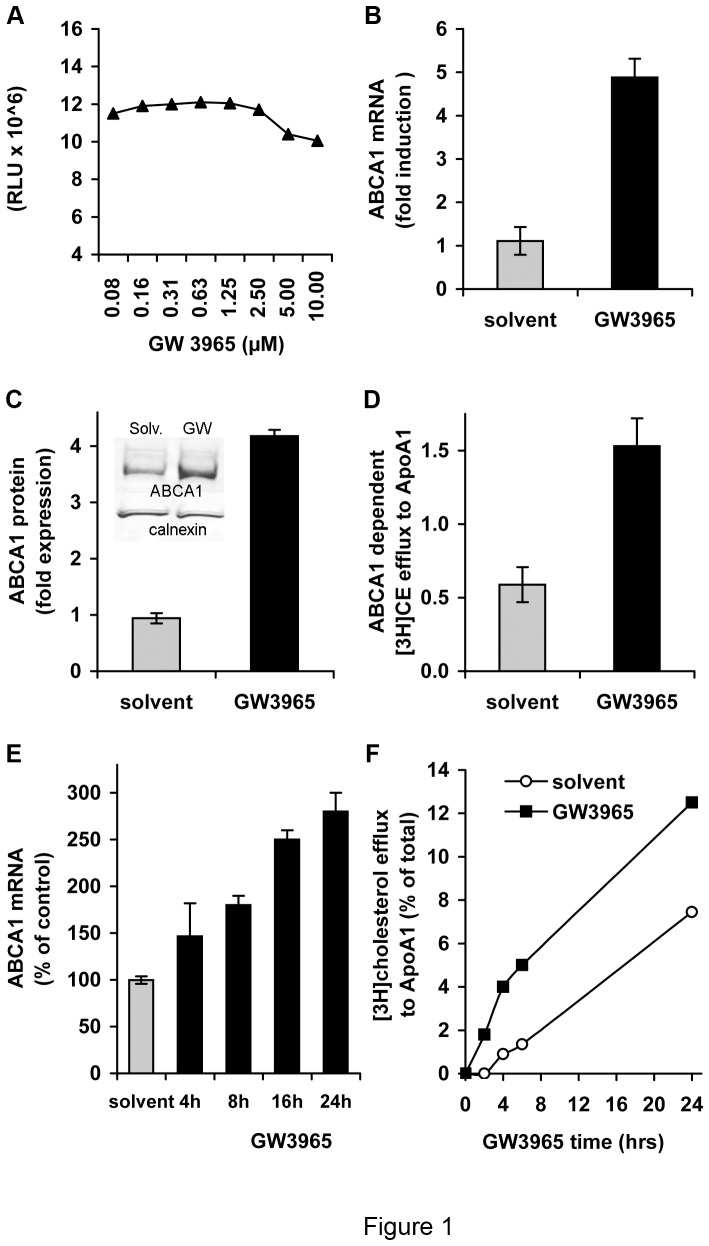
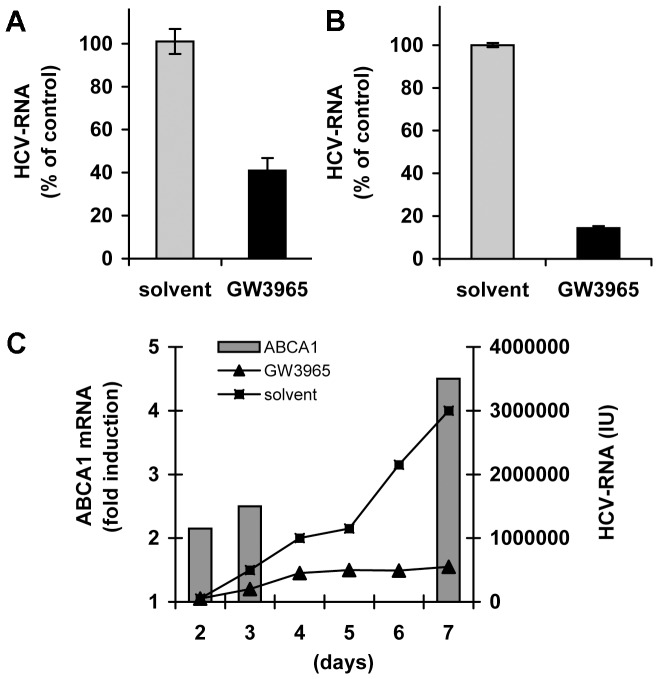
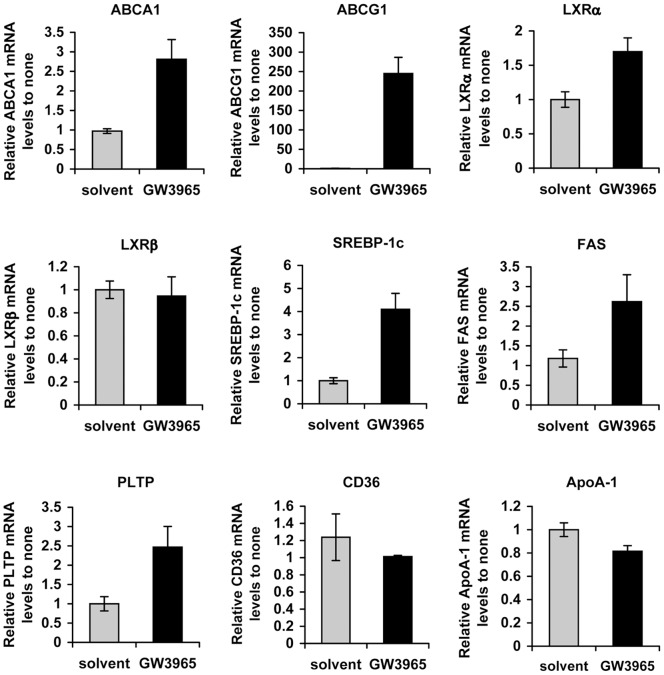
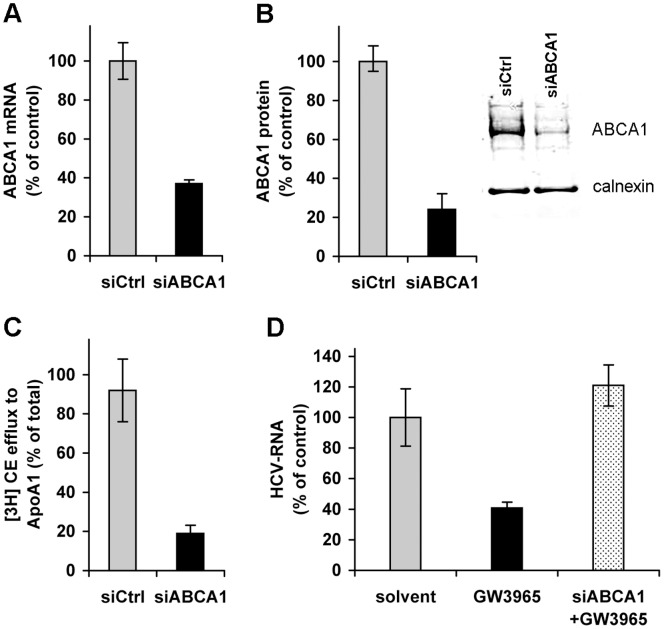
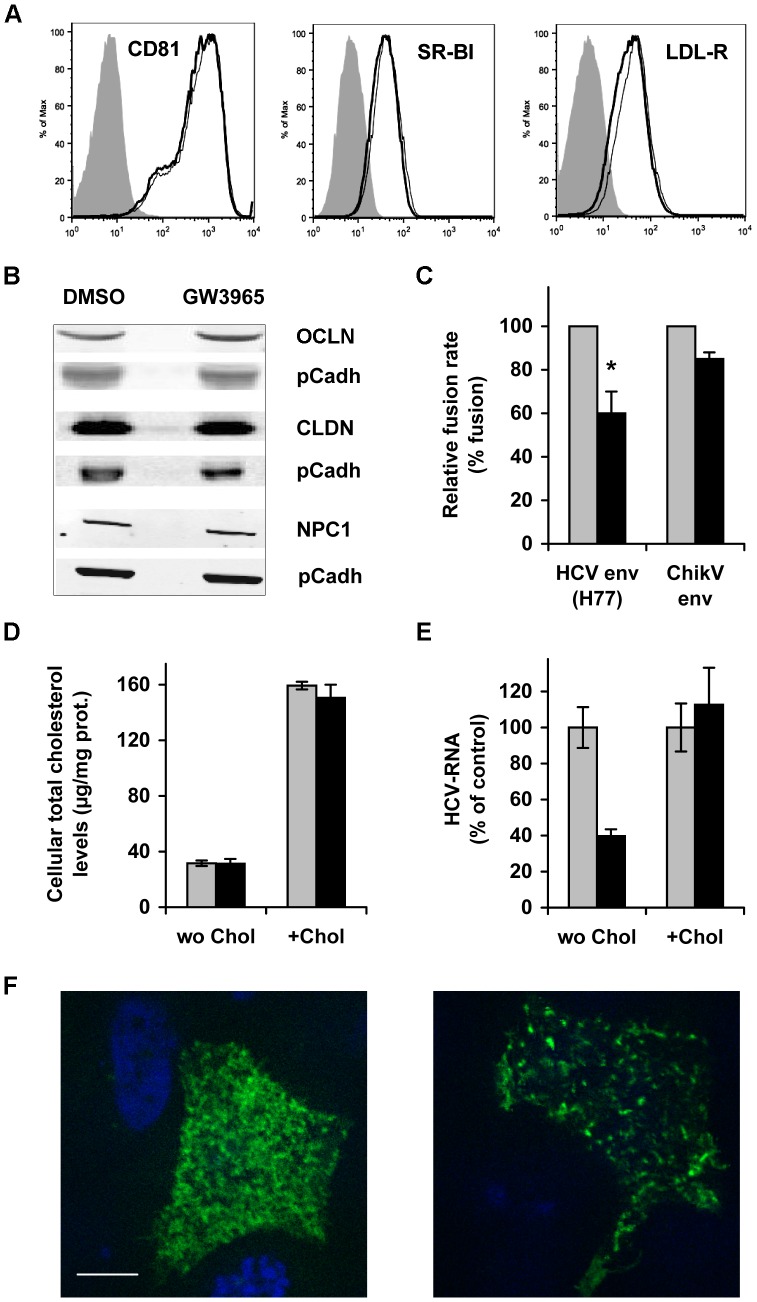
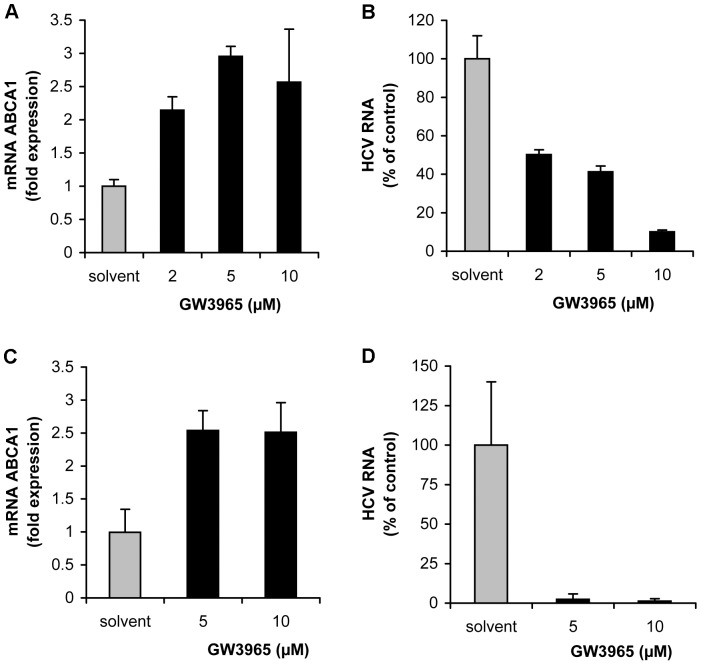
Hepatitis C virus (HCV) establishes infection using host lipid metabolism pathways that are thus considered potential targets for indirect anti-HCV strategies. HCV enters the cell via clathrin-dependent endocytosis, interacting with several receptors, and virus-cell fusion, which depends on acidic pH and the integrity of cholesterol-rich domains of the hepatocyte membrane. The ATP-binding Cassette Transporter A1 (ABCA1) mediates cholesterol efflux from hepatocytes to extracellular Apolipoprotein A1 and moves cholesterol within cell membranes. Furthermore, it generates high-density lipoprotein (HDL) particles. HDL protects against arteriosclerosis and cardiovascular disease. We show that the up-regulation of ABCA1 gene expression and its cholesterol efflux function in Huh7.5 hepatoma cells, using the liver X receptor (LXR) agonist GW3965, impairs HCV infection and decreases levels of virus produced. ABCA1-stimulation inhibited HCV cell entry, acting on virus-host cell fusion, but had no impact on virus attachment, replication, or assembly/secretion. It did not affect infectivity or properties of virus particles produced. Silencing of the ABCA1 gene and reduction of the specific cholesterol efflux function counteracted the inhibitory effect of the GW3965 on HCV infection, providing evidence for a key role of ABCA1 in this process. Impaired virus-cell entry correlated with the reorganisation of cholesterol-rich membrane microdomains (lipid rafts). The inhibitory effect could be reversed by an exogenous cholesterol supply, indicating that restriction of HCV infection was induced by changes of cholesterol content/distribution in membrane regions essential for virus-cell fusion. Stimulation of ABCA1 expression by GW3965 inhibited HCV infection of both human primary hepatocytes and isolated human liver slices. This study reveals that pharmacological stimulation of the ABCA1-dependent cholesterol efflux pathway disrupts membrane cholesterol homeostasis, leading to the inhibition of virus-cell fusion and thus HCV cell entry. Therefore besides other beneficial roles, ABCA1 might represent a potential target for HCV therapy.
Conflict of interest statement
Figures
Similar articles
-
LXR driven induction of HDL-cholesterol is independent of intestinal cholesterol absorption and ABCA1 protein expression.Lipids. 2014 Jan;49(1):71-83. doi: 10.1007/s11745-013-3853-8. Epub 2013 Oct 27. Lipids. 2014. PMID: 24163219
-
Extracellular lipid-free apolipoprotein E inhibits HCV replication and induces ABCG1-dependent cholesterol efflux.Gut. 2017 May;66(5):896-907. doi: 10.1136/gutjnl-2015-311289. Epub 2016 Sep 8. Gut. 2017. PMID: 27609828 Free PMC article.
-
Interdependent Impact of Lipoprotein Receptors and Lipid-Lowering Drugs on HCV Infectivity.Cells. 2021 Jun 29;10(7):1626. doi: 10.3390/cells10071626. Cells. 2021. PMID: 34209751 Free PMC article.
-
Lipids: a key for hepatitis C virus entry and a potential target for antiviral strategies.Biochimie. 2013 Jan;95(1):96-102. doi: 10.1016/j.biochi.2012.07.016. Epub 2012 Aug 1. Biochimie. 2013. PMID: 22884392 Review.
-
The Role of the ATP-Binding Cassette A1 (ABCA1) in Human Disease.Int J Mol Sci. 2021 Feb 5;22(4):1593. doi: 10.3390/ijms22041593. Int J Mol Sci. 2021. PMID: 33562440 Free PMC article. Review.
Cited by
-
Differential expression of microRNA, miR-150 and enhancer of zeste homolog 2 (EZH2) in peripheral blood cells as early prognostic markers of severe forms of dengue.J Biomed Sci. 2020 Jan 18;27(1):25. doi: 10.1186/s12929-020-0620-z. J Biomed Sci. 2020. PMID: 31954402 Free PMC article.
-
Intracellular and Plasma Membrane Events in Cholesterol Transport and Homeostasis.J Lipids. 2018 Aug 6;2018:3965054. doi: 10.1155/2018/3965054. eCollection 2018. J Lipids. 2018. PMID: 30174957 Free PMC article. Review.
-
Lipids in Pathophysiology and Development of the Membrane Lipid Therapy: New Bioactive Lipids.Membranes (Basel). 2021 Nov 24;11(12):919. doi: 10.3390/membranes11120919. Membranes (Basel). 2021. PMID: 34940418 Free PMC article. Review.
-
Chronic hepatitis C virus infection and lipoprotein metabolism.World J Gastroenterol. 2015 Sep 28;21(36):10299-313. doi: 10.3748/wjg.v21.i36.10299. World J Gastroenterol. 2015. PMID: 26420957 Free PMC article. Review.
-
Global analysis of HBV-mediated host proteome and ubiquitylome change in HepG2.2.15 human hepatoblastoma cell line.Cell Biosci. 2021 Apr 17;11(1):75. doi: 10.1186/s13578-021-00588-3. Cell Biosci. 2021. PMID: 33865438 Free PMC article.
References
-
- Asselah T, Marcellin P (2012) Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver International 32: 88–102. - PubMed
-
- Buhler S, Bartenschlager R (2012) New targets for antiviral therapy of chronic hepatitis C. Liver Int. 32 Suppl 19–16. - PubMed
-
- Bartenschlager R, Cosset FL, Lohmann V (2010) Hepatitis C virus replication cycle. J Hepatol 53: 583–585. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
