

Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy
- PMID: 24590288
- PMCID: PMC3973095
- DOI: 10.1172/JCI71318
Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy
Abstract
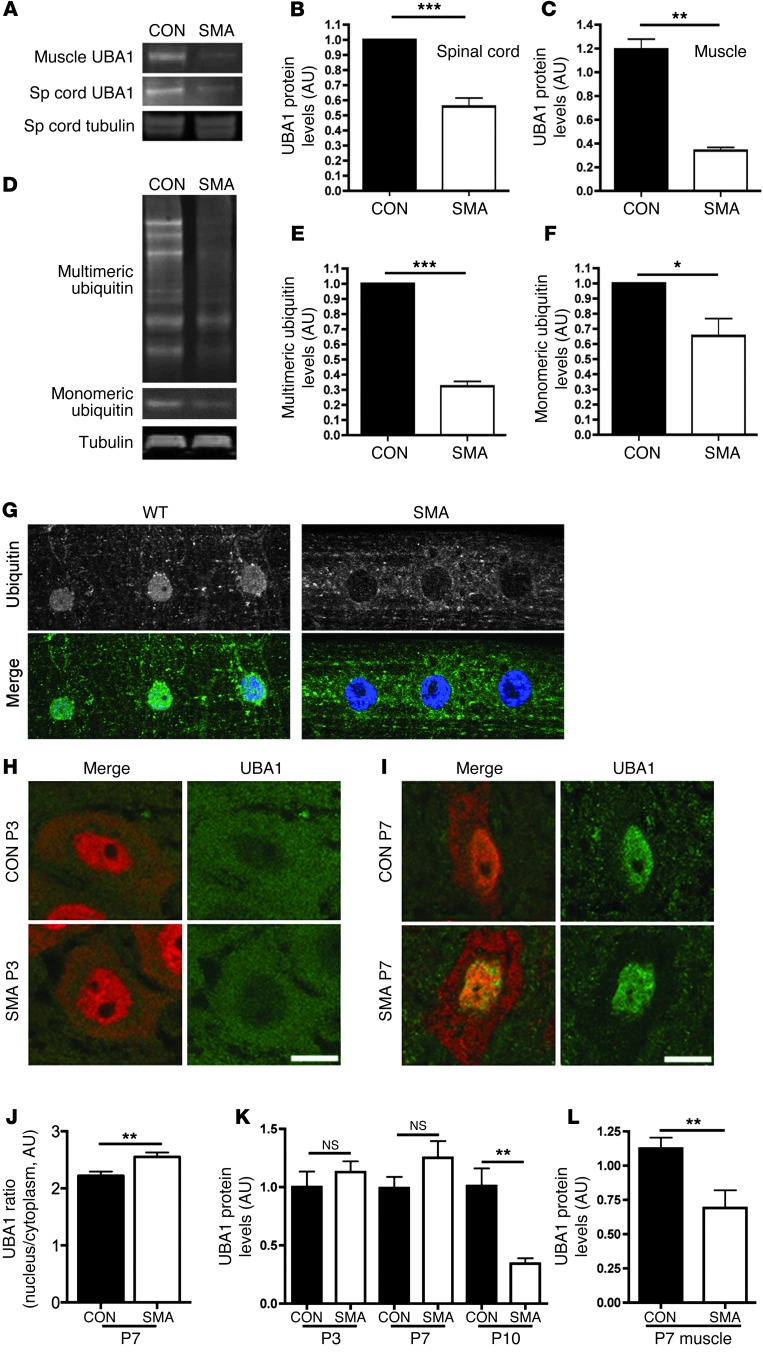
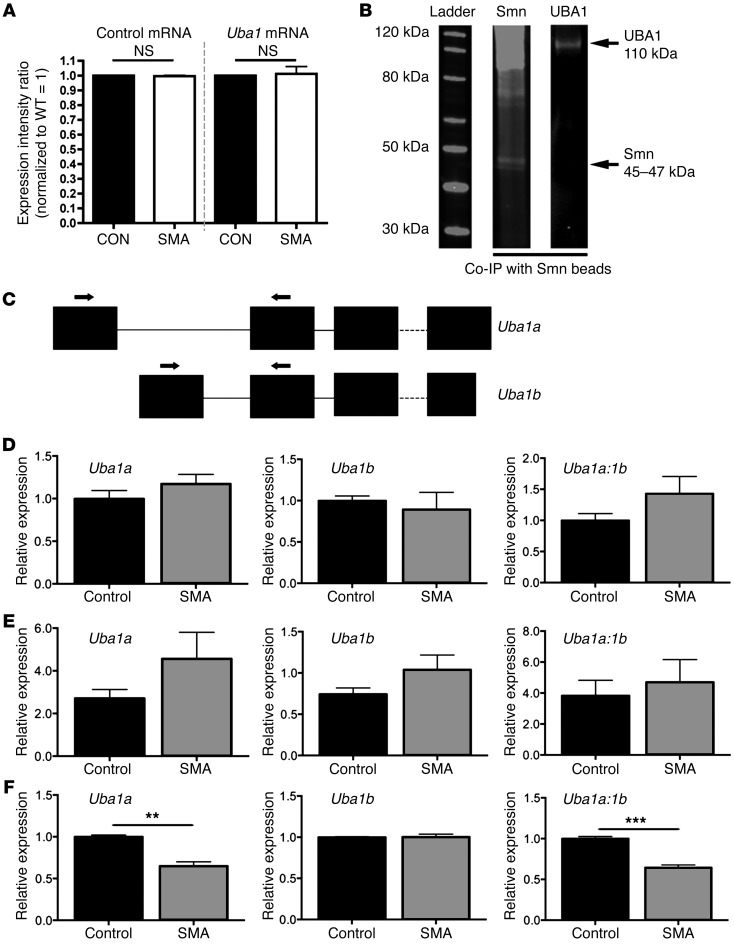
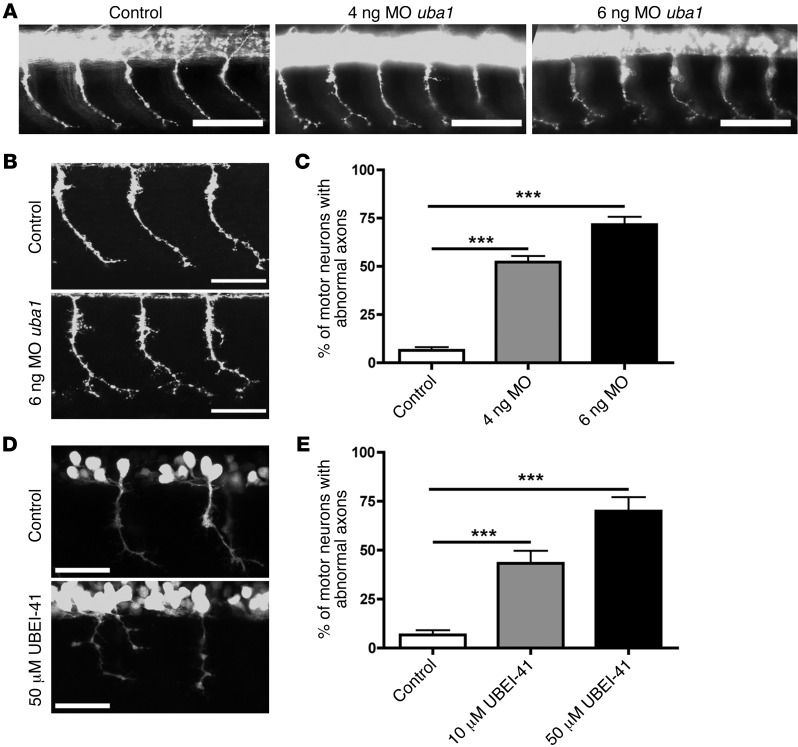
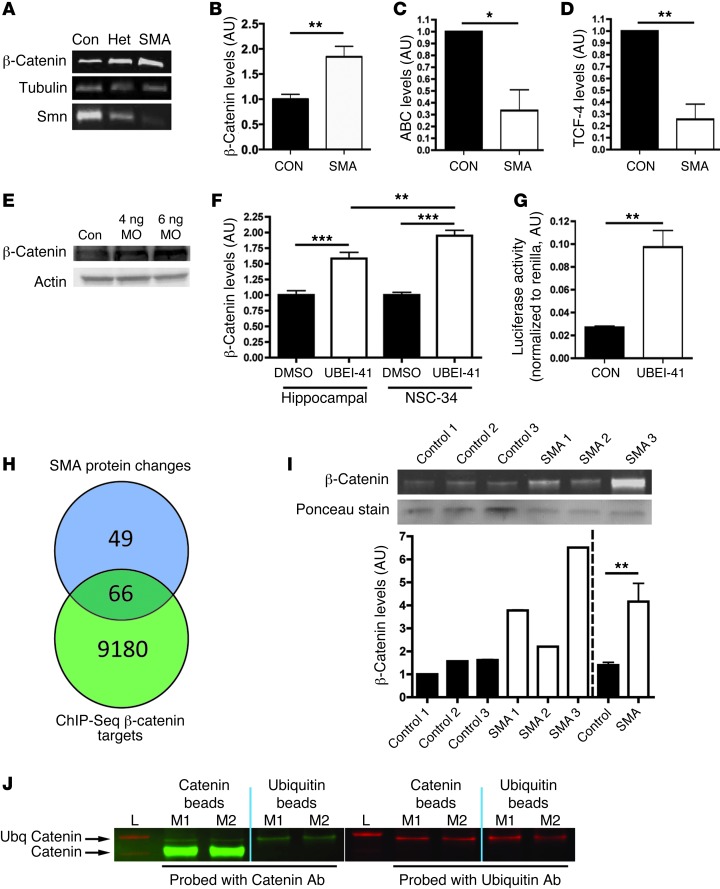
The autosomal recessive neurodegenerative disease spinal muscular atrophy (SMA) results from low levels of survival motor neuron (SMN) protein; however, it is unclear how reduced SMN promotes SMA development. Here, we determined that ubiquitin-dependent pathways regulate neuromuscular pathology in SMA. Using mouse models of SMA, we observed widespread perturbations in ubiquitin homeostasis, including reduced levels of ubiquitin-like modifier activating enzyme 1 (UBA1). SMN physically interacted with UBA1 in neurons, and disruption of Uba1 mRNA splicing was observed in the spinal cords of SMA mice exhibiting disease symptoms. Pharmacological or genetic suppression of UBA1 was sufficient to recapitulate an SMA-like neuromuscular pathology in zebrafish, suggesting that UBA1 directly contributes to disease pathogenesis. Dysregulation of UBA1 and subsequent ubiquitination pathways led to β-catenin accumulation, and pharmacological inhibition of β-catenin robustly ameliorated neuromuscular pathology in zebrafish, Drosophila, and mouse models of SMA. UBA1-associated disruption of β-catenin was restricted to the neuromuscular system in SMA mice; therefore, pharmacological inhibition of β-catenin in these animals failed to prevent systemic pathology in peripheral tissues and organs, indicating fundamental molecular differences between neuromuscular and systemic SMA pathology. Our data indicate that SMA-associated reduction of UBA1 contributes to neuromuscular pathogenesis through disruption of ubiquitin homeostasis and subsequent β-catenin signaling, highlighting ubiquitin homeostasis and β-catenin as potential therapeutic targets for SMA.
Figures
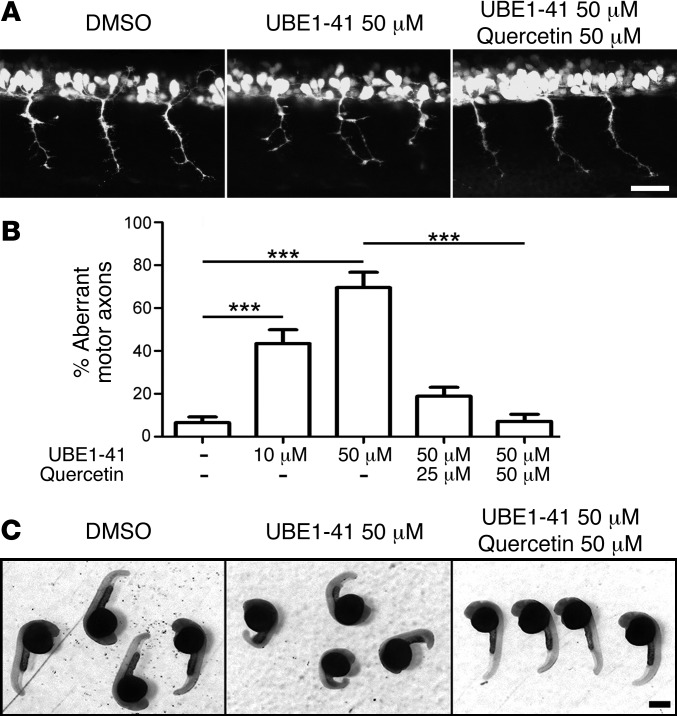
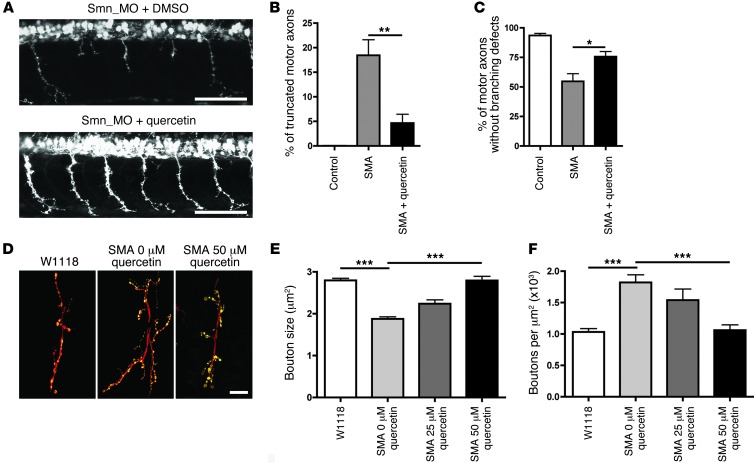

Similar articles
-
Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy.JCI Insight. 2016 Jul 21;1(11):e87908. doi: 10.1172/jci.insight.87908. JCI Insight. 2016. PMID: 27699224 Free PMC article.
-
Increased levels of UCHL1 are a compensatory response to disrupted ubiquitin homeostasis in spinal muscular atrophy and do not represent a viable therapeutic target.Neuropathol Appl Neurobiol. 2014 Dec;40(7):873-87. doi: 10.1111/nan.12168. Neuropathol Appl Neurobiol. 2014. PMID: 25041530
-
Label-free quantitative proteomic profiling identifies disruption of ubiquitin homeostasis as a key driver of Schwann cell defects in spinal muscular atrophy.J Proteome Res. 2014 Nov 7;13(11):4546-57. doi: 10.1021/pr500492j. Epub 2014 Aug 29. J Proteome Res. 2014. PMID: 25151848
-
Ubiquitination Insight from Spinal Muscular Atrophy-From Pathogenesis to Therapy: A Muscle Perspective.Int J Mol Sci. 2024 Aug 13;25(16):8800. doi: 10.3390/ijms25168800. Int J Mol Sci. 2024. PMID: 39201486 Free PMC article. Review.
-
The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration.Int J Biochem Cell Biol. 2020 Jun;123:105746. doi: 10.1016/j.biocel.2020.105746. Epub 2020 Apr 18. Int J Biochem Cell Biol. 2020. PMID: 32315770 Review.
Cited by
-
Automated in vivo drug screen in zebrafish identifies synapse-stabilising drugs with relevance to spinal muscular atrophy.Dis Model Mech. 2021 Apr 1;14(4):dmm047761. doi: 10.1242/dmm.047761. Epub 2021 Apr 26. Dis Model Mech. 2021. PMID: 33973627 Free PMC article.
-
Profilin2 regulates actin rod assembly in neuronal cells.Sci Rep. 2021 May 13;11(1):10287. doi: 10.1038/s41598-021-89397-9. Sci Rep. 2021. PMID: 33986363 Free PMC article.
-
Motor neuron biology and disease: A current perspective on infantile-onset spinal muscular atrophy.Future Neurol. 2018 Aug;13(3):161-172. doi: 10.2217/fnl-2018-0008. Epub 2018 Jul 6. Future Neurol. 2018. PMID: 31396020 Free PMC article.
-
Multi-Study Proteomic and Bioinformatic Identification of Molecular Overlap between Amyotrophic Lateral Sclerosis (ALS) and Spinal Muscular Atrophy (SMA).Brain Sci. 2018 Dec 4;8(12):212. doi: 10.3390/brainsci8120212. Brain Sci. 2018. PMID: 30518112 Free PMC article.
-
Analysis of FUS, PFN2, TDP-43, and PLS3 as potential disease severity modifiers in spinal muscular atrophy.Neurol Genet. 2019 Jan 3;6(1):e386. doi: 10.1212/NXG.0000000000000386. eCollection 2020 Feb. Neurol Genet. 2019. PMID: 32042914 Free PMC article.
References
-
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17(7):949–962. doi: 10.1093/hmg/ddm367. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
