

The role of IKKβ in Venezuelan equine encephalitis virus infection
- PMID: 24586253
- PMCID: PMC3929299
- DOI: 10.1371/journal.pone.0086745
The role of IKKβ in Venezuelan equine encephalitis virus infection
Abstract
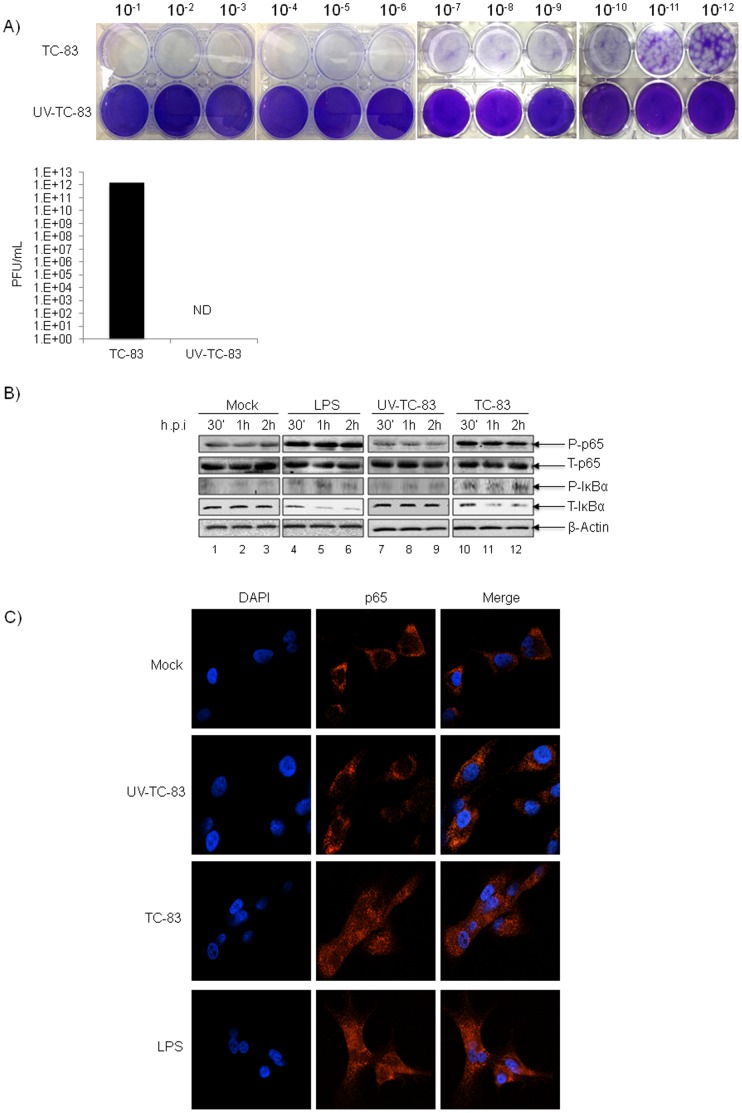
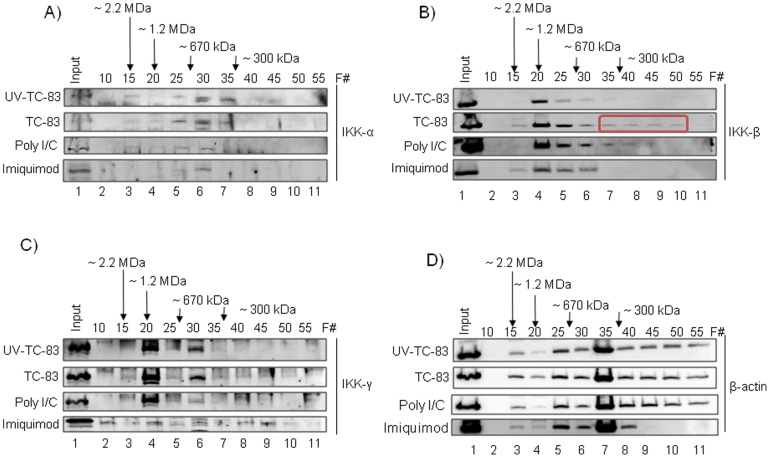
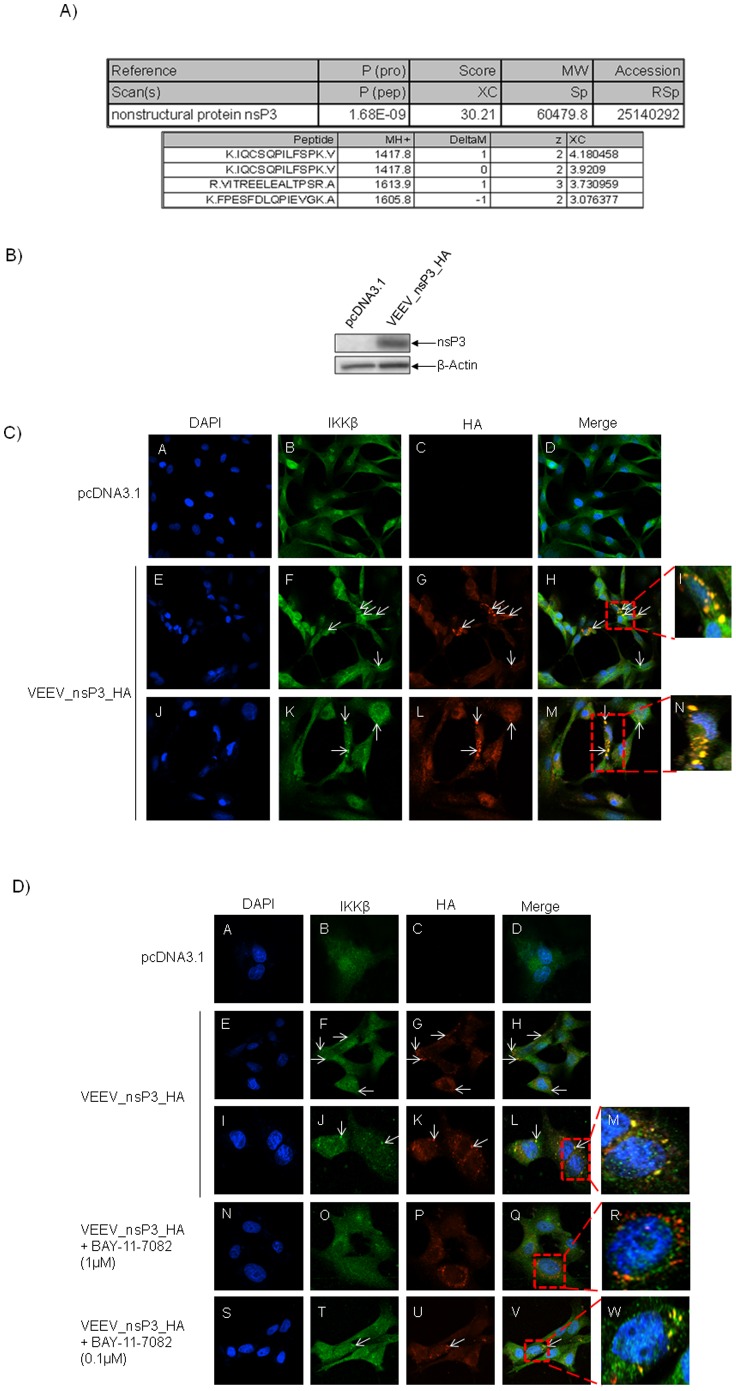
Venezuelan equine encephalitis virus (VEEV) belongs to the genus Alphavirus, family Togaviridae. VEEV infection is characterized by extensive inflammation and studies from other laboratories implicated an involvement of the NF-κB cascade in the in vivo pathology. Initial studies indicated that at early time points of VEEV infection, the NF-κB complex was activated in cells infected with the TC-83 strain of VEEV. One upstream kinase that contributes to the phosphorylation of p65 is the IKKβ component of the IKK complex. Our previous studies with Rift valley fever virus, which exhibited early activation of the NF-κB cascade in infected cells, had indicated that the IKKβ component underwent macromolecular reorganization to form a novel low molecular weight form unique to infected cells. This prompted us to investigate if the IKK complex undergoes a comparable macromolecular reorganization in VEEV infection. Size-fractionated VEEV infected cell extracts indicated a macromolecular reorganization of IKKβ in VEEV infected cells that resulted in formation of lower molecular weight complexes. Well-documented inhibitors of IKKβ function, BAY-11-7082, BAY-11-7085 and IKK2 compound IV, were employed to determine whether IKKβ function was required for the production of infectious progeny virus. A decrease in infectious viral particles and viral RNA copies was observed with inhibitor treatment in the attenuated and virulent strains of VEEV infection. In order to further validate the requirement of IKKβ for VEEV replication, we over-expressed IKKβ in cells and observed an increase in viral titers. In contrast, studies carried out using IKKβ(-/-) cells demonstrated a decrease in VEEV replication. In vivo studies demonstrated that inhibitor treatment of TC-83 infected mice increased their survival. Finally, proteomics studies have revealed that IKKβ may interact with the viral protein nsP3. In conclusion, our studies have revealed that the host IKKβ protein may be critically involved in VEEV replication.
Conflict of interest statement
Figures
Similar articles
-
Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development.Viruses. 2019 Feb 18;11(2):164. doi: 10.3390/v11020164. Viruses. 2019. PMID: 30781656 Free PMC article. Review.
-
Venezuelan Equine Encephalitis Virus nsP3 Phosphorylation Can Be Mediated by IKKβ Kinase Activity and Abrogation of Phosphorylation Inhibits Negative-Strand Synthesis.Viruses. 2020 Sep 13;12(9):1021. doi: 10.3390/v12091021. Viruses. 2020. PMID: 32933112 Free PMC article.
-
Small molecule inhibitors of Ago2 decrease Venezuelan equine encephalitis virus replication.Antiviral Res. 2014 Dec;112:26-37. doi: 10.1016/j.antiviral.2014.10.002. Epub 2014 Oct 18. Antiviral Res. 2014. PMID: 25448087
-
The ubiquitin proteasome system plays a role in venezuelan equine encephalitis virus infection.PLoS One. 2015 Apr 30;10(4):e0124792. doi: 10.1371/journal.pone.0124792. eCollection 2015. PLoS One. 2015. PMID: 25927990 Free PMC article.
-
Venezuelan Equine Encephalitis Virus Capsid-The Clever Caper.Viruses. 2017 Sep 29;9(10):279. doi: 10.3390/v9100279. Viruses. 2017. PMID: 28961161 Free PMC article. Review.
Cited by
-
The Putative Roles and Functions of Indel, Repetition and Duplication Events in Alphavirus Non-Structural Protein 3 Hypervariable Domain (nsP3 HVD) in Evolution, Viability and Re-Emergence.Viruses. 2021 May 28;13(6):1021. doi: 10.3390/v13061021. Viruses. 2021. PMID: 34071712 Free PMC article. Review.
-
Neural-Cell-Intrinsic NF-κB Signaling Enhances Reovirus Virulence.J Virol. 2023 Jan 31;97(1):e0144222. doi: 10.1128/jvi.01442-22. Epub 2022 Dec 21. J Virol. 2023. PMID: 36541803 Free PMC article.
-
NF-κB Activation Promotes Alphavirus Replication in Mature Neurons.J Virol. 2019 Nov 26;93(24):e01071-19. doi: 10.1128/JVI.01071-19. Print 2019 Dec 15. J Virol. 2019. PMID: 31554691 Free PMC article.
-
Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development.Viruses. 2019 Feb 18;11(2):164. doi: 10.3390/v11020164. Viruses. 2019. PMID: 30781656 Free PMC article. Review.
-
Inhibition of host extracellular signal-regulated kinase (ERK) activation decreases new world alphavirus multiplication in infected cells.Virology. 2014 Nov;468-470:490-503. doi: 10.1016/j.virol.2014.09.005. Epub 2014 Sep 27. Virology. 2014. PMID: 25261871 Free PMC article.
References
-
- Steele KE, Twenhafel NA (2010) REVIEW PAPER: pathology of animal models of alphavirus encephalitis. Vet Pathol 47: 790–805. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
