

Assessing the impact of human genome annotation choice on RNA-seq expression estimates
- PMID: 24564364
- PMCID: PMC3816316
- DOI: 10.1186/1471-2105-14-S11-S8
Assessing the impact of human genome annotation choice on RNA-seq expression estimates
Abstract
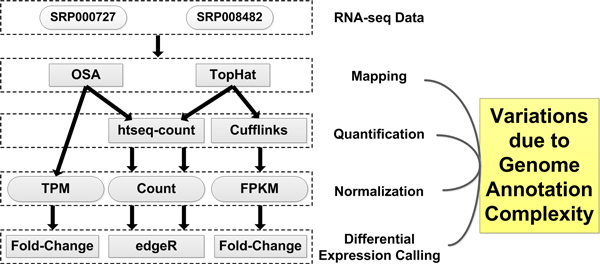
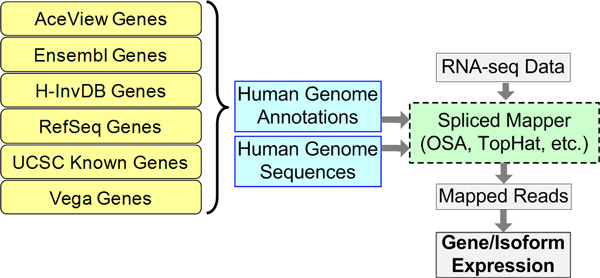
Background: Genome annotation is a crucial component of RNA-seq data analysis. Much effort has been devoted to producing an accurate and rational annotation of the human genome. An annotated genome provides a comprehensive catalogue of genomic functional elements. Currently, at least six human genome annotations are publicly available, including AceView Genes, Ensembl Genes, H-InvDB Genes, RefSeq Genes, UCSC Known Genes, and Vega Genes. Characteristics of these annotations differ because of variations in annotation strategies and information sources. When performing RNA-seq data analysis, researchers need to choose a genome annotation. However, the effect of genome annotation choice on downstream RNA-seq expression estimates is still unclear. This study (1) investigates the effect of different genome annotations on RNA-seq quantification and (2) provides guidelines for choosing a genome annotation based on research focus.
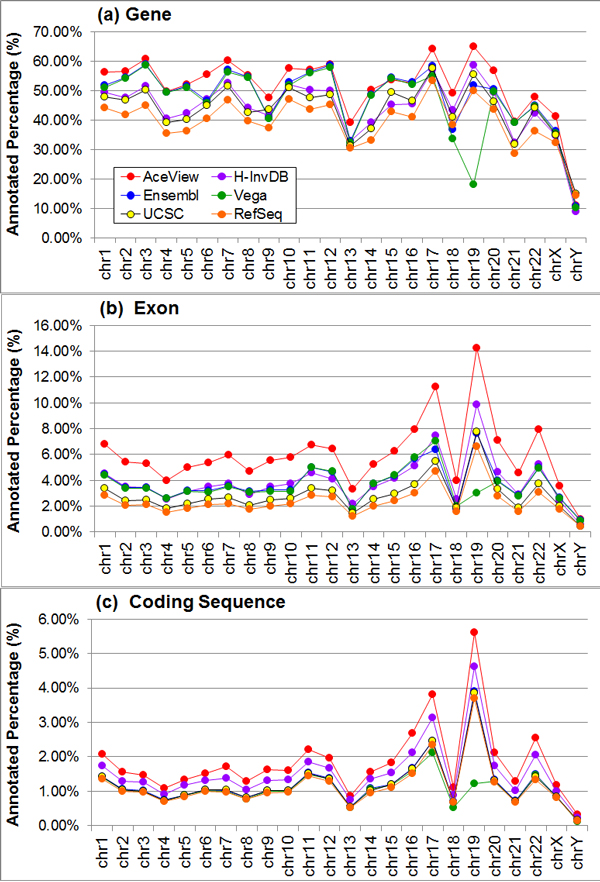
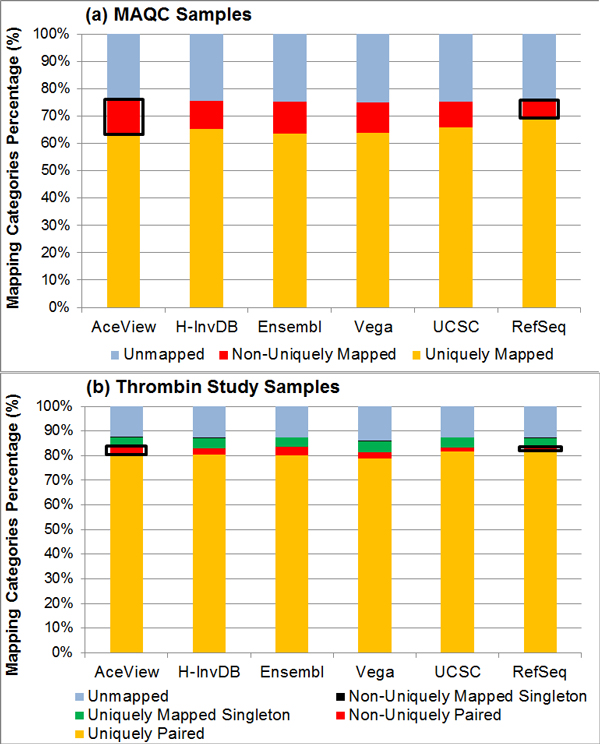
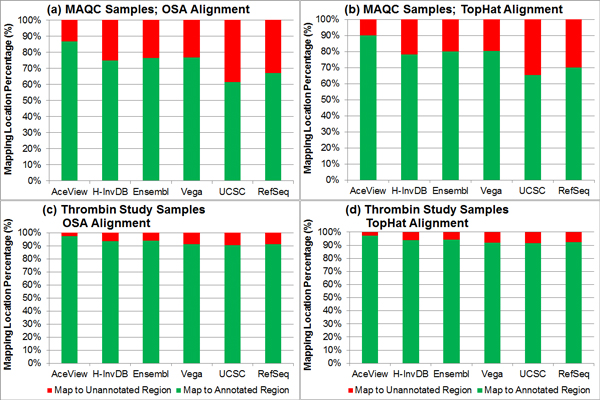
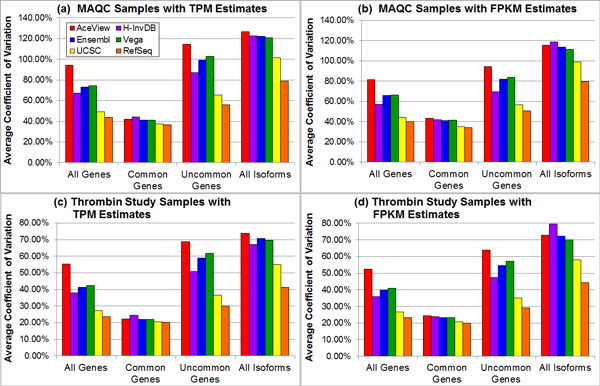
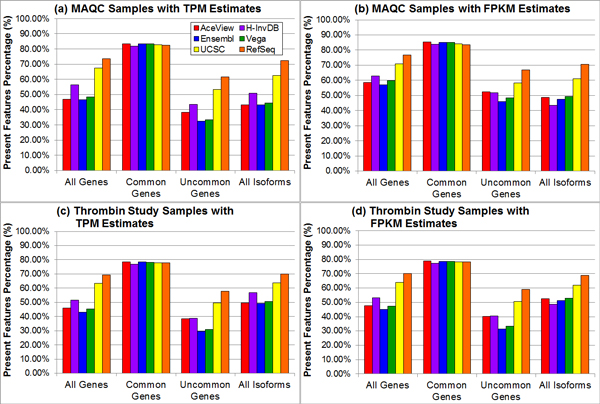
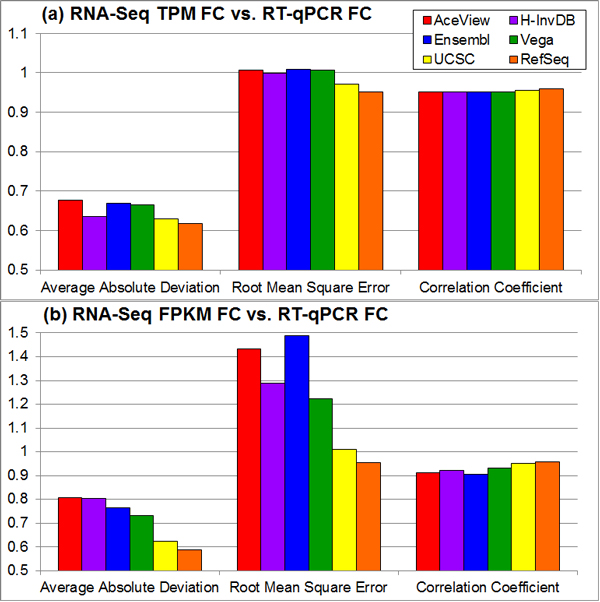
Results: We define the complexity of human genome annotations in terms of the number of genes, isoforms, and exons. This definition facilitates an investigation of potential relationships between complexity and variations in RNA-seq quantification. We apply several evaluation metrics to demonstrate the impact of genome annotation choice on RNA-seq expression estimates. In the mapping stage, the least complex genome annotation, RefSeq Genes, appears to have the highest percentage of uniquely mapped short sequence reads. In the quantification stage, RefSeq Genes results in the most stable expression estimates in terms of the average coefficient of variation over all genes. Stable expression estimates in the quantification stage translate to accurate statistics for detecting differentially expressed genes. We observe that RefSeq Genes produces the most accurate fold-change measures with respect to a ground truth of RT-qPCR gene expression estimates.
Conclusions: Based on the observed variations in the mapping, quantification, and differential expression calling stages, we demonstrate that the selection of human genome annotation results in different gene expression estimates. When conducting research that emphasizes reproducible and robust gene expression estimates, a less complex genome annotation may be preferred. However, simpler genome annotations may limit opportunities for identifying or characterizing novel transcriptional or regulatory mechanisms. When conducting research that aims to be more exploratory, a more complex genome annotation may be preferred.
Figures
Similar articles
-
A comprehensive evaluation of ensembl, RefSeq, and UCSC annotations in the context of RNA-seq read mapping and gene quantification.BMC Genomics. 2015 Feb 18;16(1):97. doi: 10.1186/s12864-015-1308-8. BMC Genomics. 2015. PMID: 25765860 Free PMC article.
-
Impact of gene annotation choice on the quantification of RNA-seq data.BMC Bioinformatics. 2022 Mar 30;23(1):107. doi: 10.1186/s12859-022-04644-8. BMC Bioinformatics. 2022. PMID: 35354358 Free PMC article.
-
GASS: genome structural annotation for Eukaryotes based on species similarity.BMC Genomics. 2015 Mar 4;16(1):150. doi: 10.1186/s12864-015-1353-3. BMC Genomics. 2015. PMID: 25764973 Free PMC article.
-
Characterizing and annotating the genome using RNA-seq data.Sci China Life Sci. 2017 Feb;60(2):116-125. doi: 10.1007/s11427-015-0349-4. Epub 2016 Jun 13. Sci China Life Sci. 2017. PMID: 27294835 Review.
-
An Experimental Approach to Genome Annotation: This report is based on a colloquium sponsored by the American Academy of Microbiology held July 19-20, 2004, in Washington, DC.Washington (DC): American Society for Microbiology; 2004. Washington (DC): American Society for Microbiology; 2004. PMID: 33001599 Free Books & Documents. Review.
Cited by
-
Comparison of stranded and non-stranded RNA-seq transcriptome profiling and investigation of gene overlap.BMC Genomics. 2015 Sep 3;16(1):675. doi: 10.1186/s12864-015-1876-7. BMC Genomics. 2015. PMID: 26334759 Free PMC article.
-
Use of semantic workflows to enhance transparency and reproducibility in clinical omics.Genome Med. 2015 Jul 25;7(1):73. doi: 10.1186/s13073-015-0202-y. Genome Med. 2015. PMID: 26289940 Free PMC article.
-
Effect of low-expression gene filtering on detection of differentially expressed genes in RNA-seq data.Annu Int Conf IEEE Eng Med Biol Soc. 2015;2015:6461-4. doi: 10.1109/EMBC.2015.7319872. Annu Int Conf IEEE Eng Med Biol Soc. 2015. PMID: 26737772 Free PMC article.
-
OpenProt: a more comprehensive guide to explore eukaryotic coding potential and proteomes.Nucleic Acids Res. 2019 Jan 8;47(D1):D403-D410. doi: 10.1093/nar/gky936. Nucleic Acids Res. 2019. PMID: 30299502 Free PMC article.
-
Bookend: precise transcript reconstruction with end-guided assembly.Genome Biol. 2022 Jun 29;23(1):143. doi: 10.1186/s13059-022-02700-3. Genome Biol. 2022. PMID: 35768836 Free PMC article.
References
-
- Li H, Zhou H, Wang D, Qiu J, Zhou Y, Li X, Rosenfeld MG, Ding S, Fu XD. Versatile pathway-centric approach based on high-throughput sequencing to anticancer drug discovery. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(12):4609–4614. doi: 10.1073/pnas.1200305109. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
