

mTORC1/C2 and pan-HDAC inhibitors synergistically impair breast cancer growth by convergent AKT and polysome inhibiting mechanisms
- PMID: 24562770
- PMCID: PMC4318538
- DOI: 10.1007/s10549-014-2877-y
mTORC1/C2 and pan-HDAC inhibitors synergistically impair breast cancer growth by convergent AKT and polysome inhibiting mechanisms
Abstract
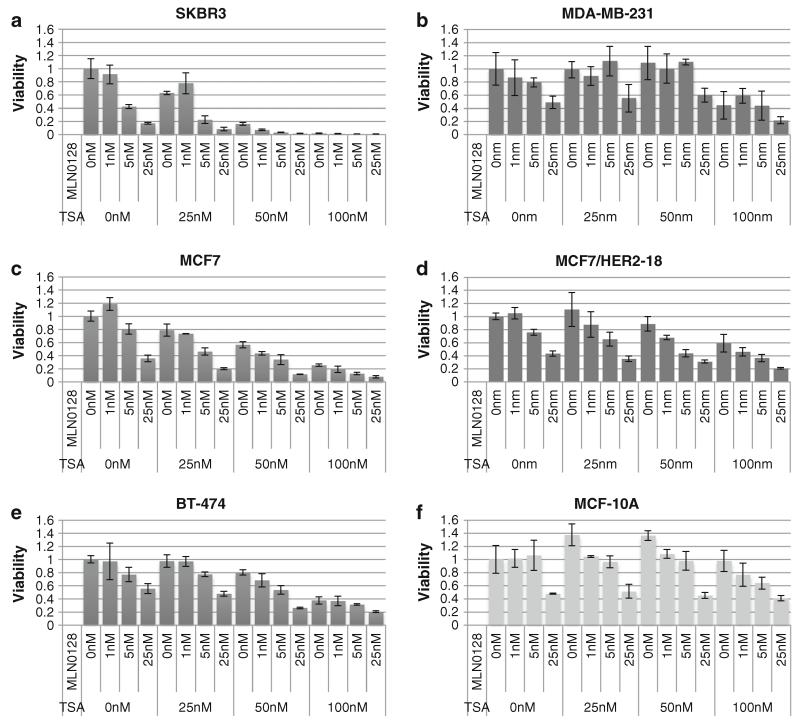
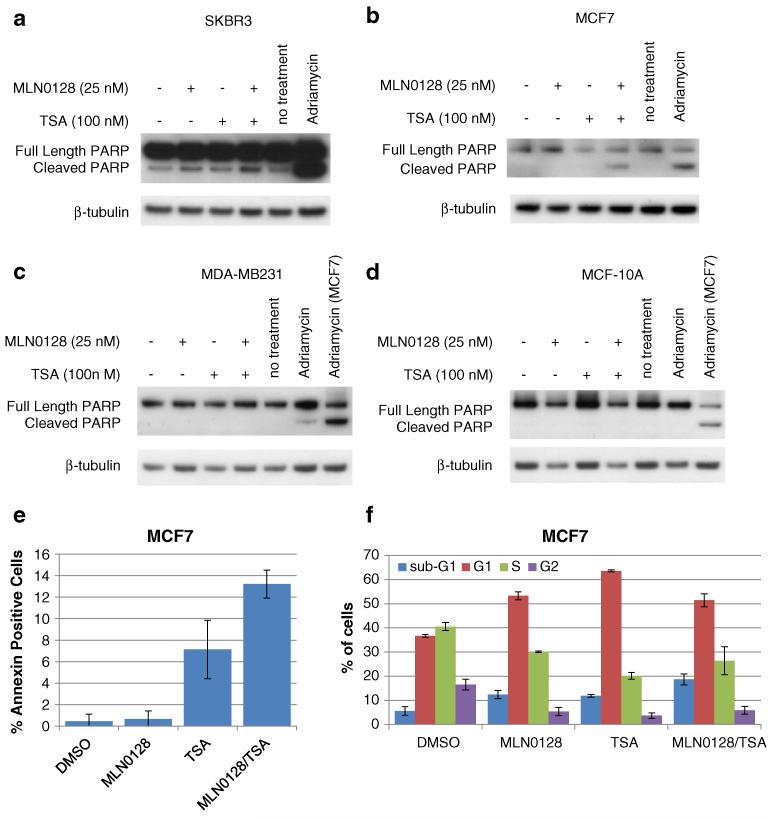
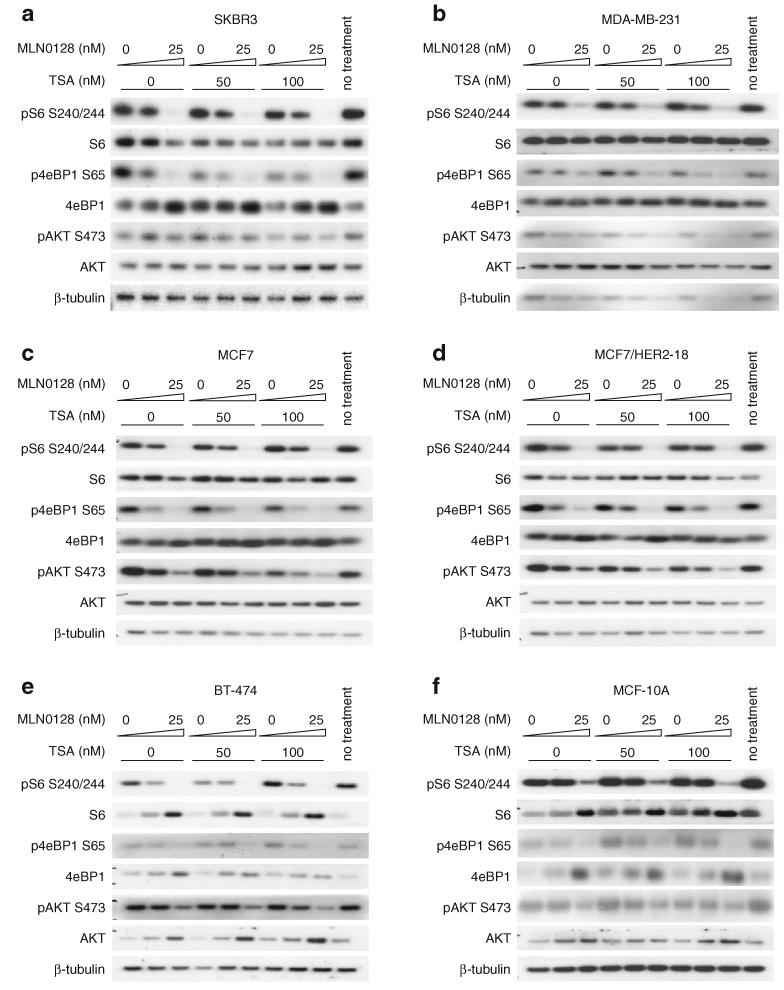
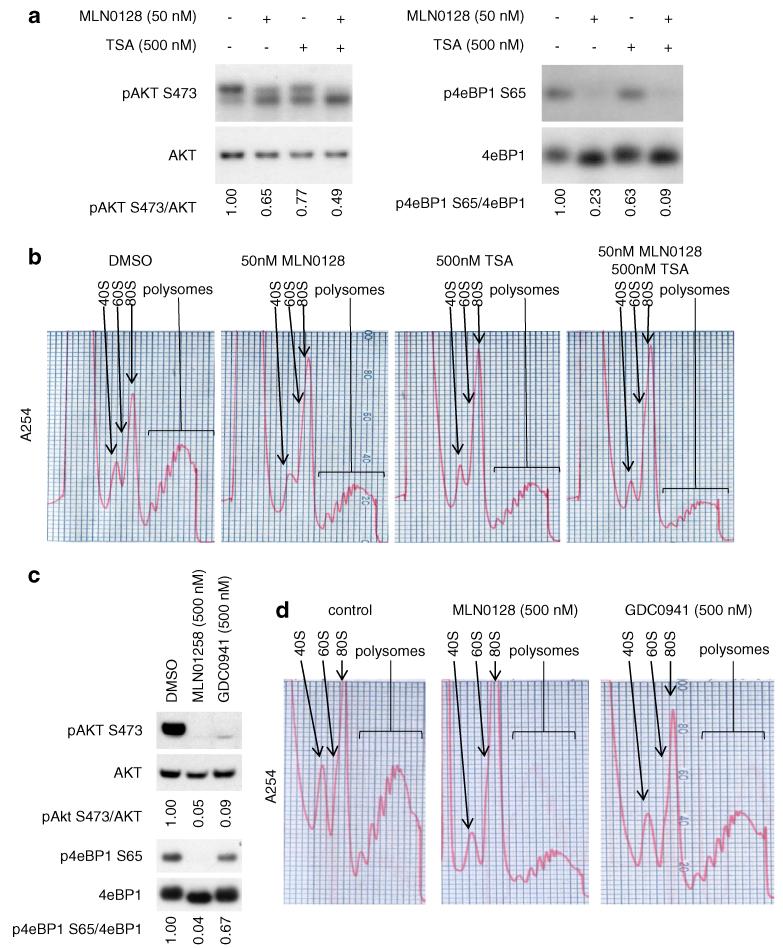
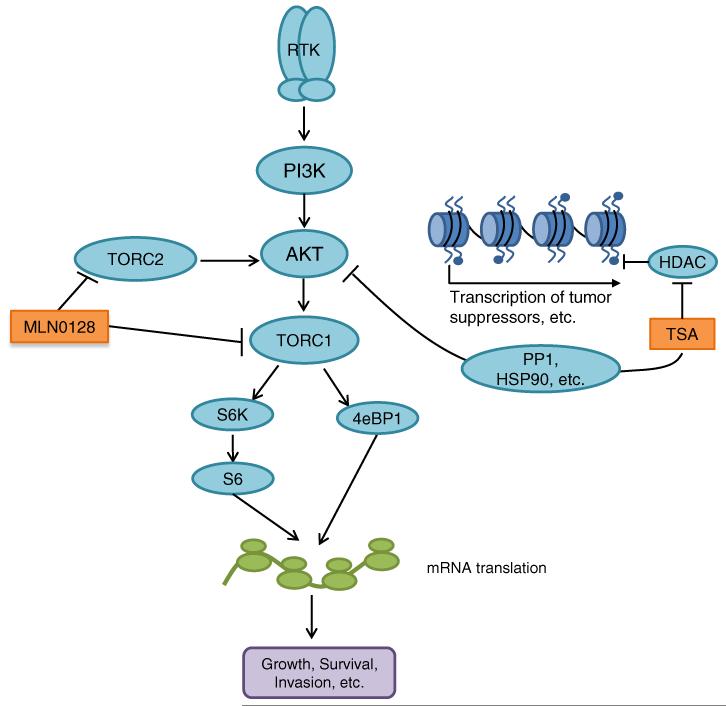
Resistance of breast cancers to targeted hormone receptor (HR) or human epidermal growth factor receptor 2 (HER2) inhibitors often occurs through dysregulation of the phosphoinositide 3-kinase, protein kinase B/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway. Presently, no targeted therapies exist for breast cancers lacking HR and HER2 overexpression, many of which also exhibit PI3K/AKT/mTOR hyper-activation. Resistance of breast cancers to current therapeutics also results, in part, from aberrant epigenetic modifications including protein acetylation regulated by histone deacetylases (HDACs). We show that the investigational drug MLN0128, which inhibits both complexes of mTOR (mTORC1 and mTORC2), and the hydroxamic acid pan-HDAC inhibitor TSA synergistically inhibit the viability of a phenotypically diverse panel of five breast cancer cell lines (HR-/+, HER2-/+). The combination of MLN0128 and TSA induces apoptosis in most breast cancer cell lines tested, but not in the non-malignant MCF-10A mammary epithelial cells. In parallel, the MLN0128/TSA combination reduces phosphorylation of AKT at S473 more than single agents alone and more so in the 5 malignant breast cancer cell lines than in the non-malignant mammary epithelial cells. Examining polysome profiles from one of the most sensitive breast cancer cell lines (SKBR3), we demonstrate that this MLN0128/TSA treatment combination synergistically impairs polysome assembly in conjunction with enhanced inhibition of 4eBP1 phosphorylation at S65. Taken together, these data indicate that the synergistic growth inhibiting consequence of combining a mTORC1/C2 inhibitor like MLN0128 with a pan-HDAC inhibitor like TSA results from their mechanistic convergence onto the PI3K/AKT/mTOR pathway, profoundly inhibiting both AKT S473 and 4eBP1 S65 phosphorylation, reducing polysome formation and cancer cell viability.
Figures
Similar articles
-
Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models.Breast Cancer Res Treat. 2012 Dec;136(3):673-82. doi: 10.1007/s10549-012-2298-8. Epub 2012 Oct 21. Breast Cancer Res Treat. 2012. PMID: 23085766
-
MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma.Mol Cancer Ther. 2015 Feb;14(2):395-406. doi: 10.1158/1535-7163.MCT-14-0711. Epub 2014 Dec 17. Mol Cancer Ther. 2015. PMID: 25519700 Free PMC article.
-
Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy.Clin Cancer Res. 2012 May 1;18(9):2603-12. doi: 10.1158/1078-0432.CCR-11-2750. Epub 2012 Mar 8. Clin Cancer Res. 2012. PMID: 22407832
-
RES-529: a PI3K/AKT/mTOR pathway inhibitor that dissociates the mTORC1 and mTORC2 complexes.Anticancer Drugs. 2016 Jul;27(6):475-87. doi: 10.1097/CAD.0000000000000354. Anticancer Drugs. 2016. PMID: 26918392 Free PMC article. Review.
-
The mTOR signalling pathway in human cancer.Int J Mol Sci. 2012;13(2):1886-1918. doi: 10.3390/ijms13021886. Epub 2012 Feb 10. Int J Mol Sci. 2012. PMID: 22408430 Free PMC article. Review.
Cited by
-
PI3K/AKT/mTOR Signaling Mediates Valproic Acid-Induced Neuronal Differentiation of Neural Stem Cells through Epigenetic Modifications.Stem Cell Reports. 2017 May 9;8(5):1256-1269. doi: 10.1016/j.stemcr.2017.04.006. Stem Cell Reports. 2017. PMID: 28494938 Free PMC article.
-
Expression of the BAD pathway is a marker of triple-negative status and poor outcome.Sci Rep. 2019 Nov 25;9(1):17496. doi: 10.1038/s41598-019-53695-0. Sci Rep. 2019. PMID: 31767884 Free PMC article.
-
mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy.Int J Mol Sci. 2019 Feb 11;20(3):755. doi: 10.3390/ijms20030755. Int J Mol Sci. 2019. PMID: 30754640 Free PMC article. Review.
-
PI3K/mTOR dual inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A synergistically exert anti-tumor activity in breast cancer.Oncotarget. 2017 Feb 14;8(7):11937-11949. doi: 10.18632/oncotarget.14442. Oncotarget. 2017. PMID: 28060760 Free PMC article.
-
Development, Maintenance, and Reversal of Multiple Drug Resistance: At the Crossroads of TFPI1, ABC Transporters, and HIF1.Cancers (Basel). 2015 Oct 16;7(4):2063-82. doi: 10.3390/cancers7040877. Cancers (Basel). 2015. PMID: 26501324 Free PMC article. Review.
References
-
- Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–6091. doi:10.1158/0008-5472.CAN-07-6854. - PMC - PubMed
-
- Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006;25(48):6384–6391. doi:10.1038/sj.onc.1209883. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
