

The role of CaMKII regulation of phospholamban activity in heart disease
- PMID: 24550830
- PMCID: PMC3913884
- DOI: 10.3389/fphar.2014.00005
The role of CaMKII regulation of phospholamban activity in heart disease
Abstract
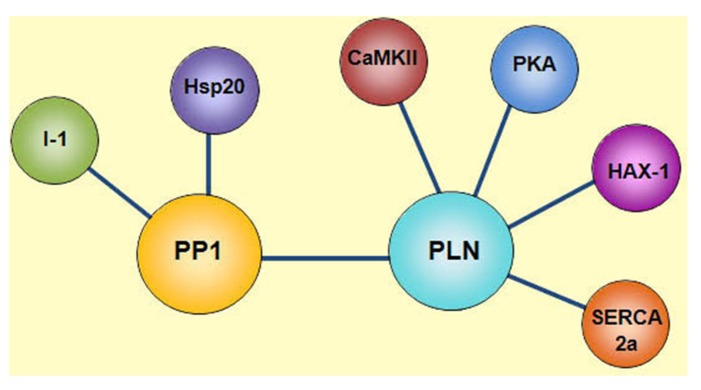
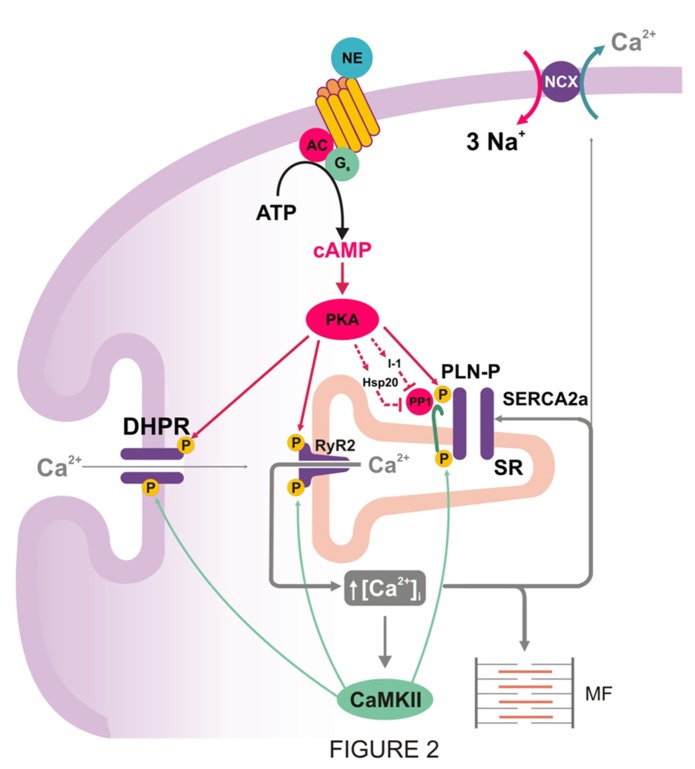
Phospholamban (PLN) is a phosphoprotein in cardiac sarcoplasmic reticulum (SR) that is a reversible regulator of the Ca(2) (+)-ATPase (SERCA2a) activity and cardiac contractility. Dephosphorylated PLN inhibits SERCA2a and PLN phosphorylation, at either Ser(16) by PKA or Thr(17) by Ca(2) (+)-calmodulin-dependent protein kinase (CaMKII), reverses this inhibition. Through this mechanism, PLN is a key modulator of SR Ca(2) (+) uptake, Ca(2) (+) load, contractility, and relaxation. PLN phosphorylation is also the main determinant of β1-adrenergic responses in the heart. Although phosphorylation of Thr(17) by CaMKII contributes to this effect, its role is subordinate to the PKA-dependent increase in cytosolic Ca(2) (+), necessary to activate CaMKII. Furthermore, the effects of PLN and its phosphorylation on cardiac function are subject to additional regulation by its interacting partners, the anti-apoptotic HAX-1 protein and Gm or the anchoring unit of protein phosphatase 1. Regulation of PLN activity by this multimeric complex becomes even more important in pathological conditions, characterized by aberrant Ca(2) (+)-cycling. In this scenario, CaMKII-dependent PLN phosphorylation has been associated with protective effects in both acidosis and ischemia/reperfusion. However, the beneficial effects of increasing SR Ca(2) (+) uptake through PLN phosphorylation may be lost or even become deleterious, when these occur in association with alterations in SR Ca(2) (+) leak. Moreover, a major characteristic in human and experimental heart failure (HF) is depressed SR Ca(2) (+) uptake, associated with decreased SERCA2a levels and dephosphorylation of PLN, leading to decreased SR Ca(2) (+) load and impaired contractility. Thus, the strategy of altering SERCA2a and/or PLN levels or activity to restore perturbed SR Ca(2) (+) uptake is a potential therapeutic tool for HF treatment. We will review here the role of CaMKII-dependent phosphorylation of PLN at Thr(17) on cardiac function under physiological and pathological conditions.
Keywords: CaMKII; PLN regulation; acidosis; heart failure; ischemia/reperfusion injury; myocardium.
Figures


Similar articles
-
Phospholamban phosphorylation by CaMKII under pathophysiological conditions.Front Biosci. 2008 May 1;13:5988-6005. doi: 10.2741/3131. Front Biosci. 2008. PMID: 18508637 Review.
-
Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions.Cardiovasc Res. 2005 Dec 1;68(3):366-75. doi: 10.1016/j.cardiores.2005.08.010. Epub 2005 Oct 13. Cardiovasc Res. 2005. PMID: 16226237 Review.
-
Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814.J Physiol. 2016 Jun 1;594(11):3005-30. doi: 10.1113/JP271622. Epub 2016 Feb 2. J Physiol. 2016. PMID: 26695843 Free PMC article.
-
Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend.J Mol Cell Cardiol. 2014 Dec;77:160-7. doi: 10.1016/j.yjmcc.2014.10.005. Epub 2014 Oct 18. J Mol Cell Cardiol. 2014. PMID: 25451386 Free PMC article. Review.
-
The importance of the Thr17 residue of phospholamban as a phosphorylation site under physiological and pathological conditions.Braz J Med Biol Res. 2006 May;39(5):563-72. doi: 10.1590/s0100-879x2006000500001. Epub 2006 Apr 20. Braz J Med Biol Res. 2006. PMID: 16648892 Review.
Cited by
-
Identification of hypertrophy-modulating Cullin-RING ubiquitin ligases in primary cardiomyocytes.Front Physiol. 2023 Mar 8;14:1134339. doi: 10.3389/fphys.2023.1134339. eCollection 2023. Front Physiol. 2023. PMID: 36969608 Free PMC article.
-
Challenges and Opportunities for Therapeutic Targeting of Calmodulin Kinase II in Heart.Front Pharmacol. 2020 Feb 5;11:35. doi: 10.3389/fphar.2020.00035. eCollection 2020. Front Pharmacol. 2020. PMID: 32116711 Free PMC article. Review.
-
Autonomous activation of CaMKII exacerbates diastolic calcium leak during beta-adrenergic stimulation in cardiomyocytes of metabolic syndrome rats.Cell Calcium. 2020 Nov;91:102267. doi: 10.1016/j.ceca.2020.102267. Epub 2020 Aug 12. Cell Calcium. 2020. PMID: 32920522 Free PMC article.
-
CaMKII-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases.Am J Physiol Heart Circ Physiol. 2016 Sep 1;311(3):H532-44. doi: 10.1152/ajpheart.00765.2015. Epub 2016 Jul 8. Am J Physiol Heart Circ Physiol. 2016. PMID: 27402669 Free PMC article.
-
Perturbations of Native Membrane Protein Structure in Alkyl Phosphocholine Detergents: A Critical Assessment of NMR and Biophysical Studies.Chem Rev. 2018 Apr 11;118(7):3559-3607. doi: 10.1021/acs.chemrev.7b00570. Epub 2018 Feb 28. Chem Rev. 2018. PMID: 29488756 Free PMC article. Review.
References
-
- Bassani R., Mattiazzi A., Bers D. M. (1995). CaMK-II is responsible for activity-dependent acceleration of relaxation in intact rat ventricular myocytes. Am. J. Physiol. 268 H703–H712 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous