

Cav2.1 channels control multivesicular release by relying on their distance from exocytotic Ca2+ sensors at rat cerebellar granule cells
- PMID: 24453334
- PMCID: PMC6705314
- DOI: 10.1523/JNEUROSCI.2388-13.2014
Cav2.1 channels control multivesicular release by relying on their distance from exocytotic Ca2+ sensors at rat cerebellar granule cells
Abstract
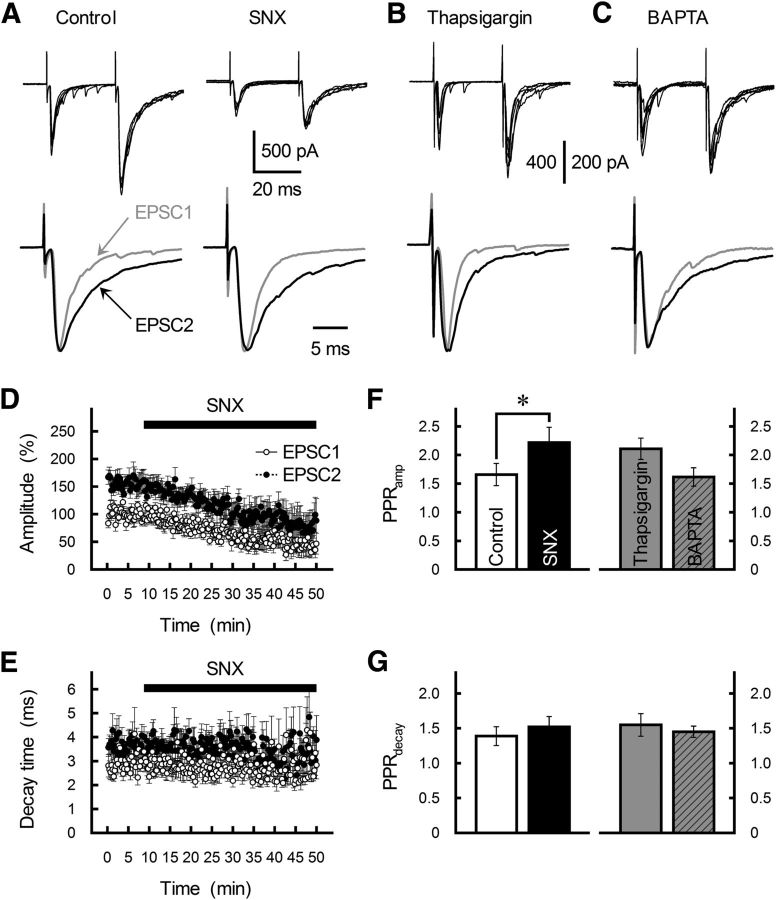
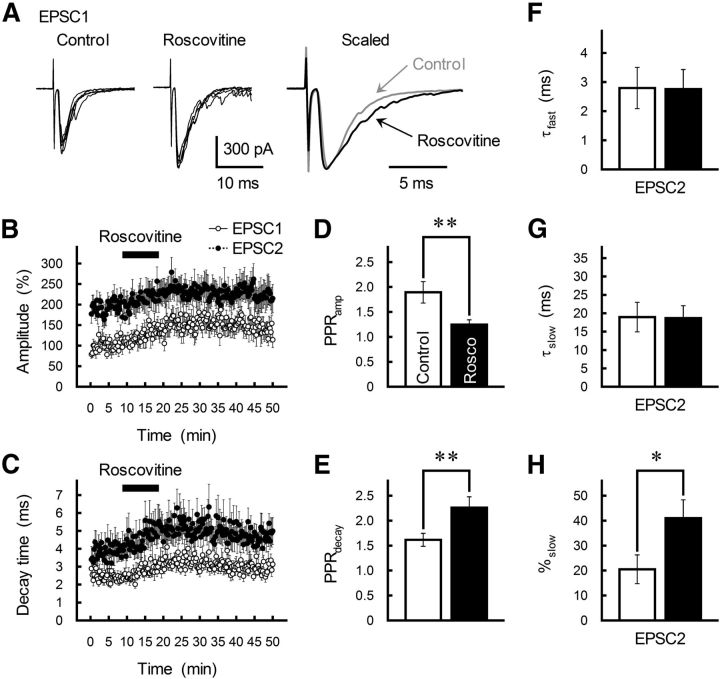
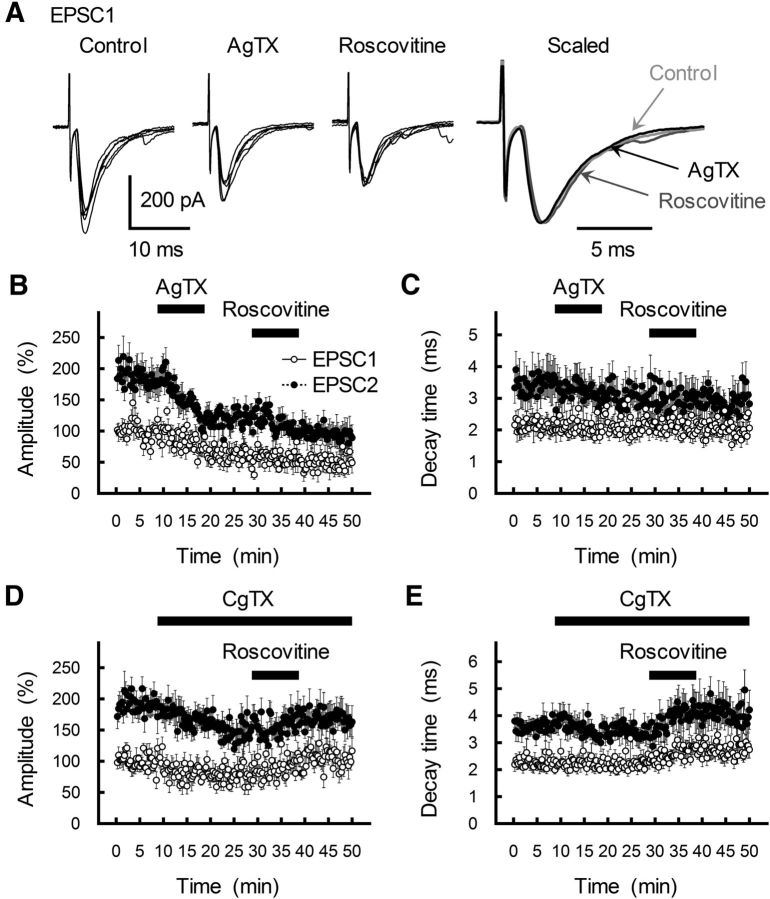
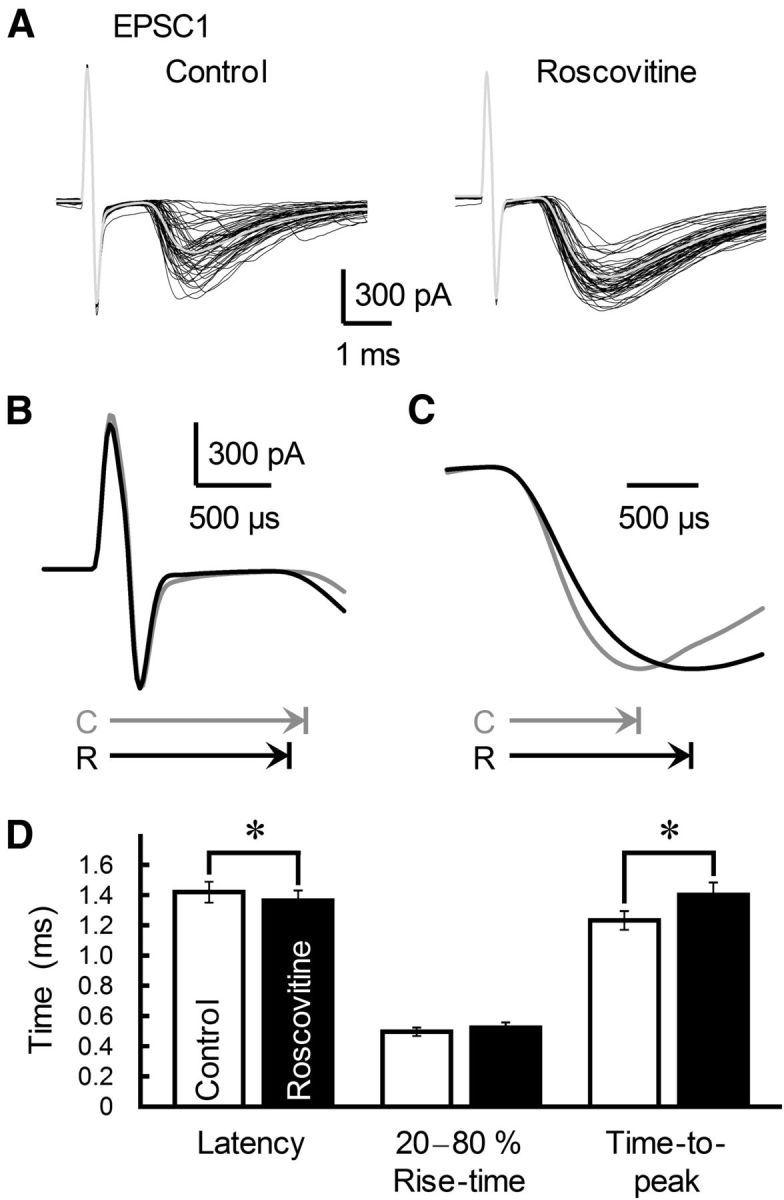
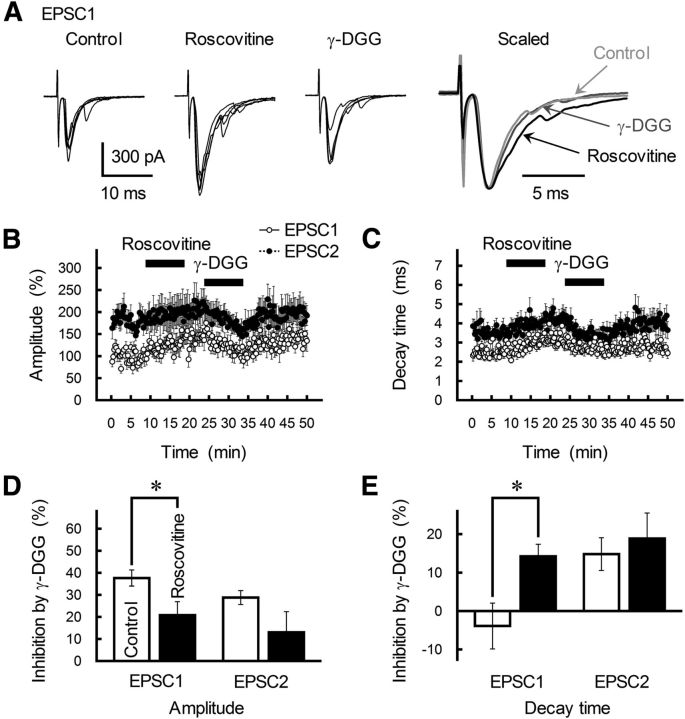
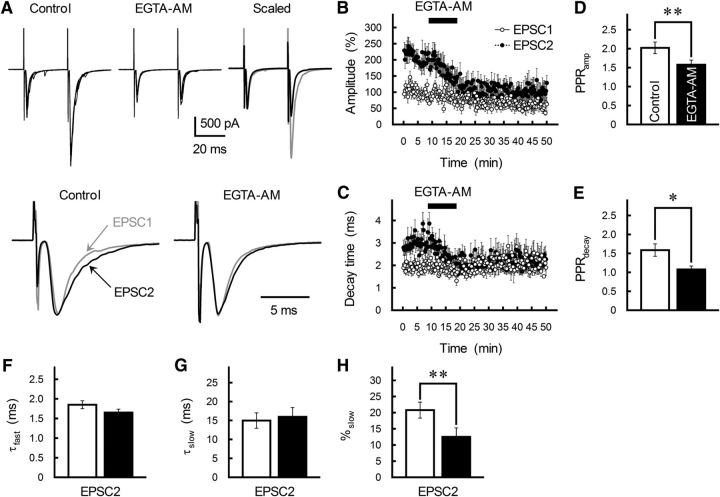
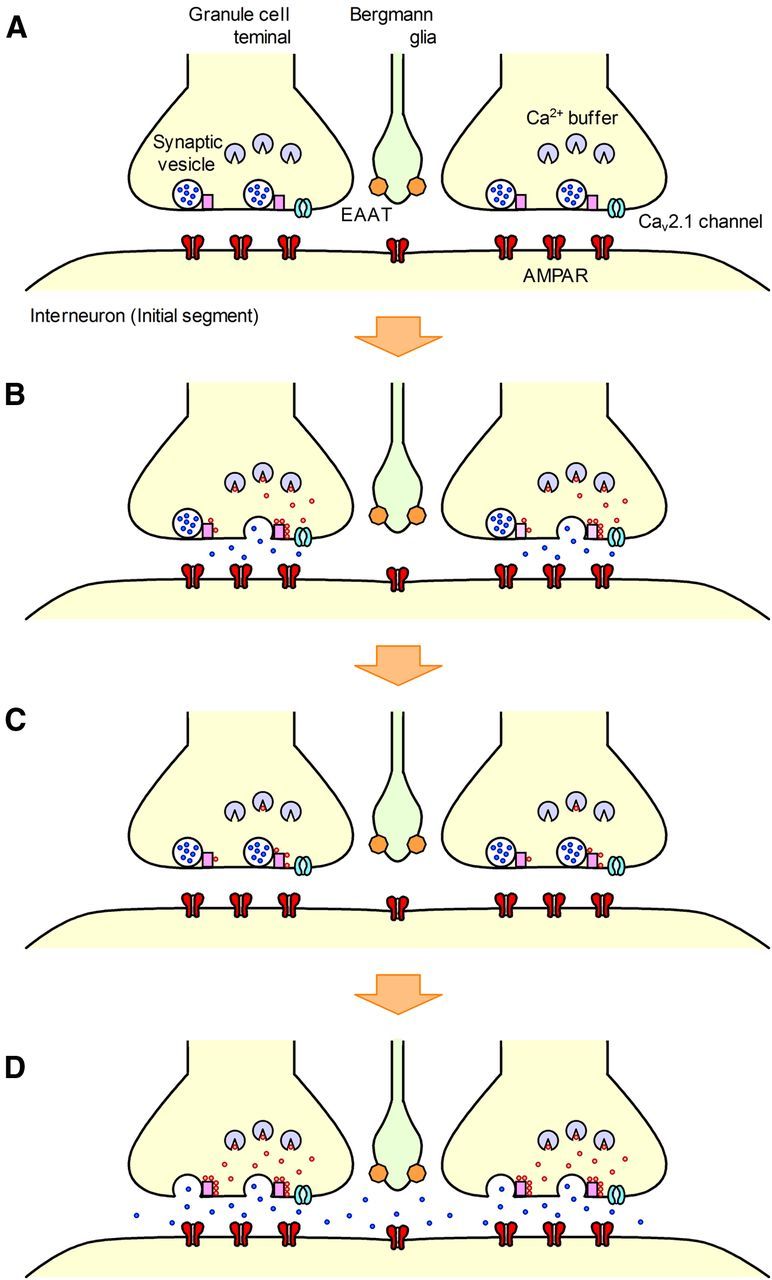
The concomitant release of multiple numbers of synaptic vesicles [multivesicular release (MVR)] in response to a single presynaptic action potential enhances the flexibility of synaptic transmission. However, the molecular mechanisms underlying MVR at a single CNS synapse remain unclear. Here, we show that the Cav2.1 subtype (P/Q-type) of the voltage-gated calcium channel is specifically responsible for the induction of MVR. In the rat cerebellar cortex, paired-pulse activation of granule cell (GC) ascending fibers leads not only to a facilitation of the peak amplitude (PPFamp) but also to a prolongation of the decay time (PPPdecay) of the EPSCs recorded from molecular layer interneurons. PPFamp is elicited by a transient increase in the number of released vesicles. PPPdecay is highly dependent on MVR and is caused by dual mechanisms: (1) a delayed release and (2) an extrasynaptic spillover of the GC transmitter glutamate and subsequent pooling of the glutamate among active synapses. PPPdecay was specifically suppressed by the Cav2.1 channel blocker ω-agatoxin IVA, while PPFamp responded to Cav2.2/Cav2.3 (N-type/R-type) channel blockers. The membrane-permeable slow Ca(2+) chelator EGTA-AM profoundly reduced the decay time constant (τdecay) of the second EPSC; however, it only had a negligible impact on that of the first, thereby eliminating PPPdecay. These results suggest that the distance between presynaptic Cav2.1 channels and exocytotic Ca(2+) sensors is a key determinant of MVR. By transducing presynaptic action potential firings into unique Ca(2+) signals and vesicle release profiles, Cav2.1 channels contribute to the encoding and processing of neural information.
Keywords: Ca2+ microdomain; roscovitine; whole-cell patch clamp.
Figures




Similar articles
-
Synaptic Multivesicular Release in the Cerebellar Cortex: Its Mechanism and Role in Neural Encoding and Processing.Cerebellum. 2016 Apr;15(2):201-7. doi: 10.1007/s12311-015-0677-5. Cerebellum. 2016. PMID: 25971904
-
Topographical distance between presynaptic Ca2+ channels and exocytotic Ca2+ sensors contributes to differential facilitatory actions of roscovitine on neurotransmitter release at cerebellar glutamatergic and GABAergic synapses.Eur J Neurosci. 2021 Nov;54(9):7048-7062. doi: 10.1111/ejn.15487. Epub 2021 Oct 21. Eur J Neurosci. 2021. PMID: 34622493
-
Roscovitine differentially facilitates cerebellar glutamatergic and GABAergic neurotransmission by enhancing Cav 2.1 channel-mediated multivesicular release.Eur J Neurosci. 2020 Aug;52(3):3002-3021. doi: 10.1111/ejn.14771. Epub 2020 Jun 27. Eur J Neurosci. 2020. PMID: 32383214
-
Presynaptic voltage-gated calcium channels in the auditory brainstem.Mol Cell Neurosci. 2021 Apr;112:103609. doi: 10.1016/j.mcn.2021.103609. Epub 2021 Mar 1. Mol Cell Neurosci. 2021. PMID: 33662542 Free PMC article. Review.
-
Presynaptic Calcium Channels.Int J Mol Sci. 2019 May 6;20(9):2217. doi: 10.3390/ijms20092217. Int J Mol Sci. 2019. PMID: 31064106 Free PMC article. Review.
Cited by
-
Synaptic Multivesicular Release in the Cerebellar Cortex: Its Mechanism and Role in Neural Encoding and Processing.Cerebellum. 2016 Apr;15(2):201-7. doi: 10.1007/s12311-015-0677-5. Cerebellum. 2016. PMID: 25971904
-
Effects of Ketamine on Neuronal Spontaneous Excitatory Postsynaptic Currents and Miniature Excitatory Postsynaptic Currents in the Somatosensory Cortex of Rats.Iran J Med Sci. 2016 Jul;41(4):275-82. Iran J Med Sci. 2016. PMID: 27365548 Free PMC article.
-
Opposite Roles in Short-Term Plasticity for N-Type and P/Q-Type Voltage-Dependent Calcium Channels in GABAergic Neuronal Connections in the Rat Cerebral Cortex.J Neurosci. 2018 Nov 14;38(46):9814-9828. doi: 10.1523/JNEUROSCI.0337-18.2018. Epub 2018 Sep 24. J Neurosci. 2018. PMID: 30249804 Free PMC article.
-
Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis.J Neurosci. 2014 Jun 11;34(24):8151-63. doi: 10.1523/JNEUROSCI.4415-13.2014. J Neurosci. 2014. PMID: 24920620 Free PMC article.
-
Single granule cells excite Golgi cells and evoke feedback inhibition in the cochlear nucleus.J Neurosci. 2015 Mar 18;35(11):4741-50. doi: 10.1523/JNEUROSCI.3665-14.2015. J Neurosci. 2015. PMID: 25788690 Free PMC article.
References
-
- Adams PJ, Rungta RL, Garcia E, van den Maagdenberg AM, MacVicar BA, Snutch TP. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc Natl Acad Sci U S A. 2010;107:18694–18699. doi: 10.1073/pnas.1009500107. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous