

Cellular senescence and its effector programs
- PMID: 24449267
- PMCID: PMC3909793
- DOI: 10.1101/gad.235184.113
Cellular senescence and its effector programs
Abstract
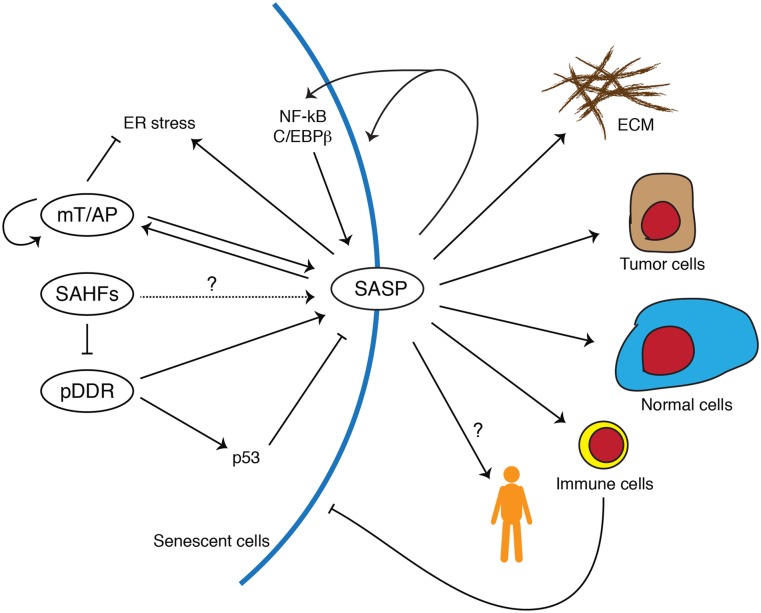
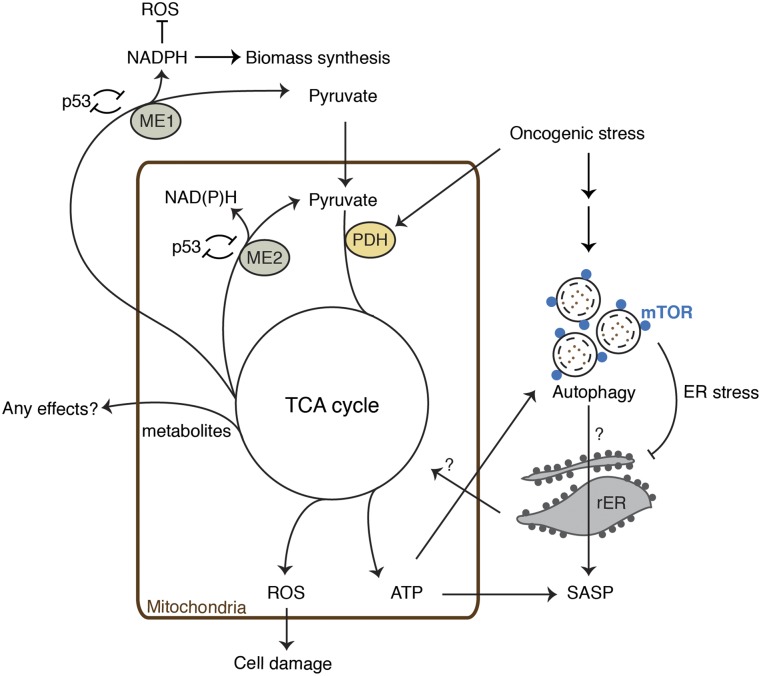
Cellular senescence is a stress response that accompanies stable exit from the cell cycle. Classically, senescence, particularly in human cells, involves the p53 and p16/Rb pathways, and often both of these tumor suppressor pathways need to be abrogated to bypass senescence. In parallel, a number of effector mechanisms of senescence have been identified and characterized. These studies suggest that senescence is a collective phenotype of these multiple effectors, and their intensity and combination can be different depending on triggers and cell types, conferring a complex and diverse nature to senescence. Series of studies on senescence-associated secretory phenotype (SASP) in particular have revealed various layers of functionality of senescent cells in vivo. Here we discuss some key features of senescence effectors and attempt to functionally link them when it is possible.
Keywords: DNA damage; aging; inflammation; metabolism; oncogenes; senescence; tumor suppressors.
Figures
Similar articles
-
Genes and pathways involved in senescence bypass identified by functional genetic screens.Mech Ageing Dev. 2021 Mar;194:111432. doi: 10.1016/j.mad.2021.111432. Epub 2021 Jan 8. Mech Ageing Dev. 2021. PMID: 33422562 Review.
-
Autophagy facilitates oncogene-induced senescence.Autophagy. 2009 Oct;5(7):1046-7. doi: 10.4161/auto.5.7.9444. Epub 2009 Oct 7. Autophagy. 2009. PMID: 19652542
-
Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence.Genes Dev. 2020 Mar 1;34(5-6):428-445. doi: 10.1101/gad.331272.119. Epub 2020 Jan 30. Genes Dev. 2020. PMID: 32001510 Free PMC article.
-
The role of chromatin reorganization in the process of cellular senescence.Curr Drug Targets. 2012 Dec;13(13):1593-602. doi: 10.2174/138945012803529983. Curr Drug Targets. 2012. PMID: 22998188 Review.
-
Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype.J Biol Chem. 2011 Oct 21;286(42):36396-403. doi: 10.1074/jbc.M111.257071. Epub 2011 Aug 31. J Biol Chem. 2011. PMID: 21880712 Free PMC article.
Cited by
-
Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype.Front Cell Dev Biol. 2021 Mar 29;9:645593. doi: 10.3389/fcell.2021.645593. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33855023 Free PMC article. Review.
-
Interphase Chromosomes in Replicative Senescence: Chromosome Positioning as a Senescence Biomarker and the Lack of Nuclear Motor-Driven Chromosome Repositioning in Senescent Cells.Front Cell Dev Biol. 2021 May 24;9:640200. doi: 10.3389/fcell.2021.640200. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34113611 Free PMC article.
-
Quercetin influences intestinal dysbacteriosis and delays alveolar epithelial cell senescence by regulating PTEN/PI3K/AKT signaling in pulmonary fibrosis.Naunyn Schmiedebergs Arch Pharmacol. 2024 Jul;397(7):4809-4822. doi: 10.1007/s00210-023-02913-8. Epub 2023 Dec 28. Naunyn Schmiedebergs Arch Pharmacol. 2024. PMID: 38153514 Free PMC article.
-
Identification and experimental verification of senescence-related gene signatures and molecular subtypes in idiopathic pulmonary arterial hypertension.Sci Rep. 2024 Sep 27;14(1):22157. doi: 10.1038/s41598-024-72979-8. Sci Rep. 2024. PMID: 39333589 Free PMC article.
-
Efficient and simple approach to in vitro culture of primary epithelial cancer cells.Biosci Rep. 2016 Dec 9;36(6):e00423. doi: 10.1042/BSR20160208. Print 2016 Dec. Biosci Rep. 2016. PMID: 27803125 Free PMC article.
References
-
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous