

STAT5 acetylation: Mechanisms and consequences for immunological control and leukemogenesis
- PMID: 24416653
- PMCID: PMC3876427
- DOI: 10.4161/jkst.26102
STAT5 acetylation: Mechanisms and consequences for immunological control and leukemogenesis
Abstract
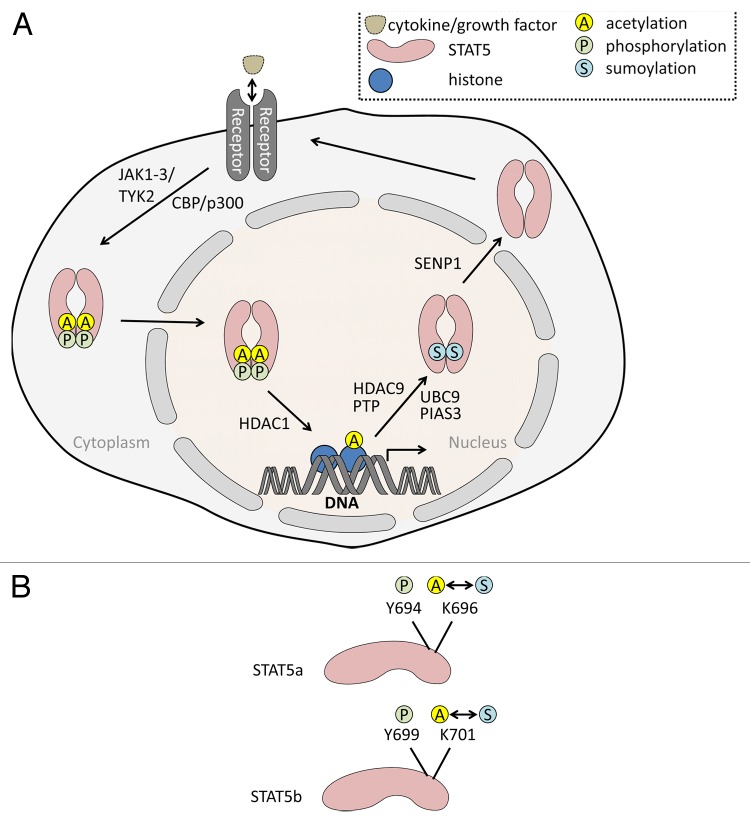
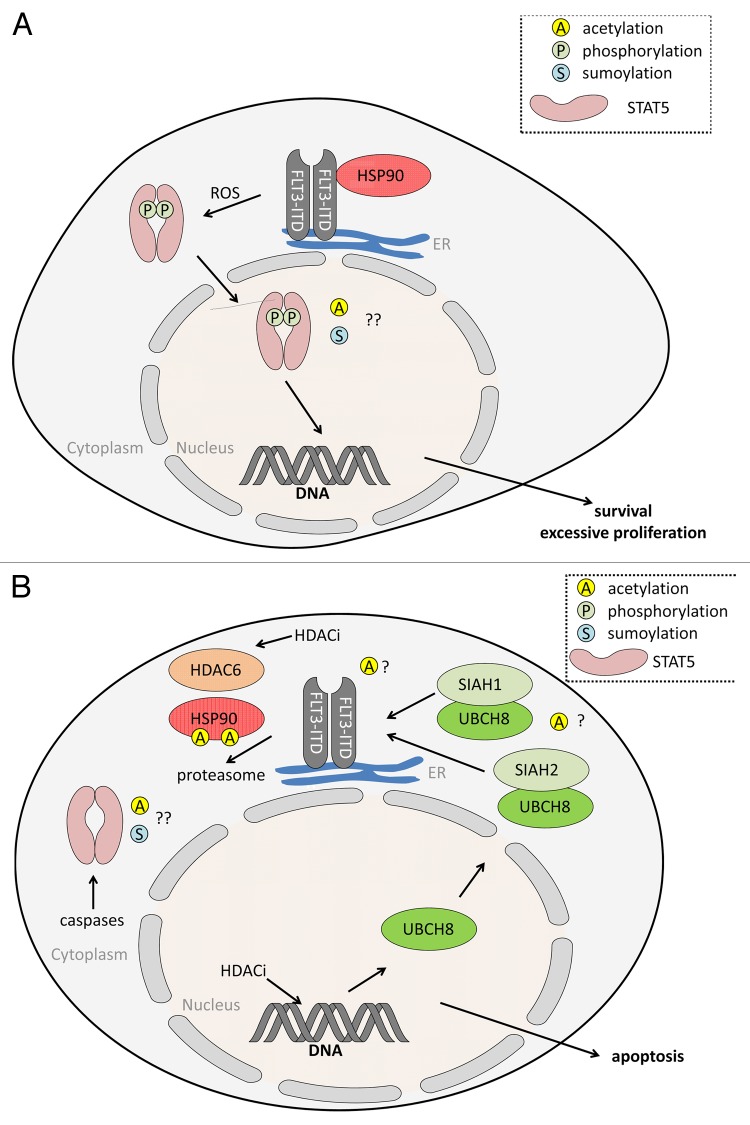
The cytokine-inducible transcription factors signal transducer and activator of transcription 5A and 5B (STAT5A and STAT5B) are important for the proper development of multicellular eukaryotes. Disturbed signaling cascades evoking uncontrolled expression of STAT5 target genes are associated with cancer and immunological failure. Here, we summarize how STAT5 acetylation is integrated into posttranslational modification networks within cells. Moreover, we focus on how inhibitors of deacetylases and tyrosine kinases can correct leukemogenic signaling nodes involving STAT5. Such small molecules can be exploited in the fight against neoplastic diseases and immunological disorders.
Keywords: HAT; HDAC; HDACi; SIAH; STAT; STAT5; UBCH8; acetylation; cancer; leukemia.
Figures
Similar articles
-
Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis.J Exp Med. 1999 Apr 19;189(8):1229-42. doi: 10.1084/jem.189.8.1229. J Exp Med. 1999. PMID: 10209040 Free PMC article.
-
Serine phosphorylation of GH-activated signal transducer and activator of transcription 5a (STAT5a) and STAT5b: impact on STAT5 transcriptional activity.Mol Endocrinol. 2001 Dec;15(12):2157-71. doi: 10.1210/mend.15.12.0746. Mol Endocrinol. 2001. PMID: 11731617
-
A sequence of the CIS gene promoter interacts preferentially with two associated STAT5A dimers: a distinct biochemical difference between STAT5A and STAT5B.Mol Cell Biol. 1998 Oct;18(10):5852-60. doi: 10.1128/MCB.18.10.5852. Mol Cell Biol. 1998. PMID: 9742102 Free PMC article.
-
The role of Stat5a and Stat5b in signaling by IL-2 family cytokines.Oncogene. 2000 May 15;19(21):2566-76. doi: 10.1038/sj.onc.1203523. Oncogene. 2000. PMID: 10851055 Review.
-
The Role of STAT5 in Tyrosine Kinase Inhibitor (IMATINIB) Resistance in CML Patients.Acta Med Indones. 2019 Oct;51(4):348-352. Acta Med Indones. 2019. PMID: 32041920 Review.
Cited by
-
Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (BET) protein function.Nucleic Acids Res. 2015 Apr 20;43(7):3524-45. doi: 10.1093/nar/gkv188. Epub 2015 Mar 13. Nucleic Acids Res. 2015. PMID: 25769527 Free PMC article.
-
STAT3 and STAT5 Targeting for Simultaneous Management of Melanoma and Autoimmune Diseases.Cancers (Basel). 2019 Sep 27;11(10):1448. doi: 10.3390/cancers11101448. Cancers (Basel). 2019. PMID: 31569642 Free PMC article. Review.
-
HDAC5 controls a hypothalamic STAT5b-TH axis, the sympathetic activation of ATP-consuming futile cycles and adult-onset obesity in male mice.Mol Metab. 2024 Dec;90:102033. doi: 10.1016/j.molmet.2024.102033. Epub 2024 Sep 19. Mol Metab. 2024. PMID: 39304061 Free PMC article.
-
Pharmacological Inhibition of Oncogenic STAT3 and STAT5 Signaling in Hematopoietic Cancers.Cancers (Basel). 2020 Jan 18;12(1):240. doi: 10.3390/cancers12010240. Cancers (Basel). 2020. PMID: 31963765 Free PMC article. Review.
-
Regulating the Regulators: The Role of Histone Deacetylase 1 (HDAC1) in Erythropoiesis.Int J Mol Sci. 2020 Nov 11;21(22):8460. doi: 10.3390/ijms21228460. Int J Mol Sci. 2020. PMID: 33187090 Free PMC article. Review.
References
-
- Ferbeyre G, Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta. 2011;1815:104–14. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous