

A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes
- PMID: 24347612
- PMCID: PMC3941102
- DOI: 10.1101/gr.164749.113
A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes
Abstract
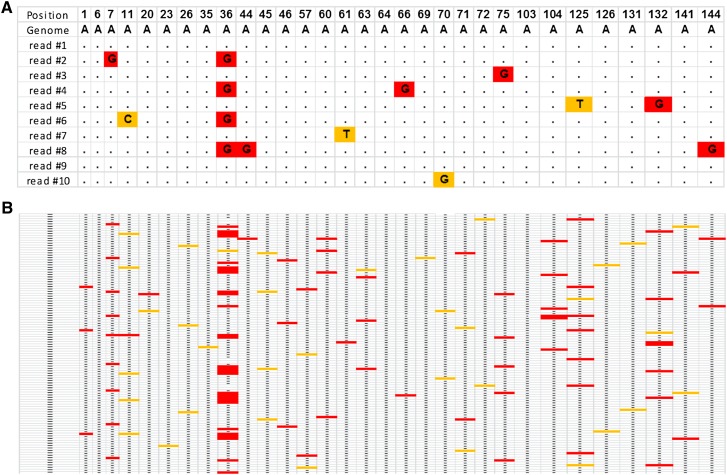
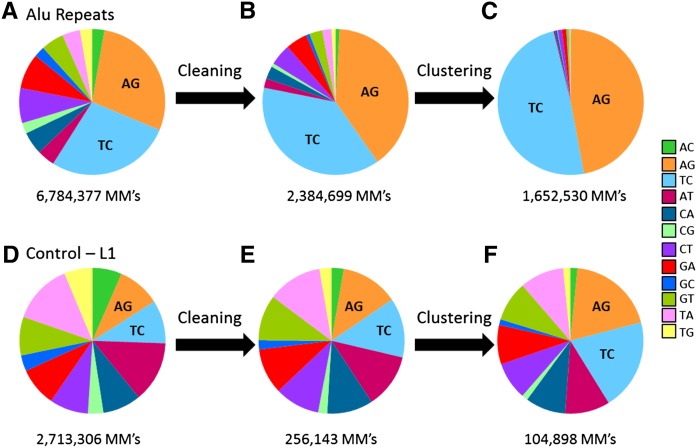
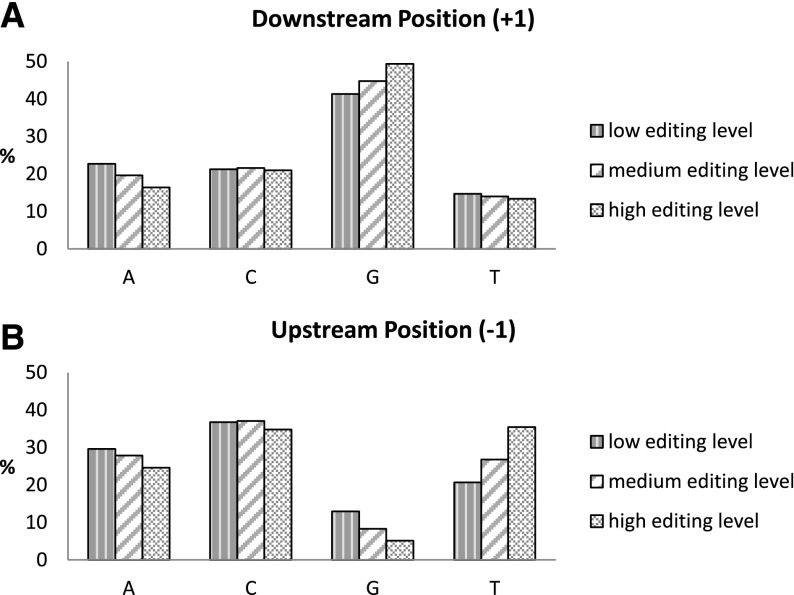
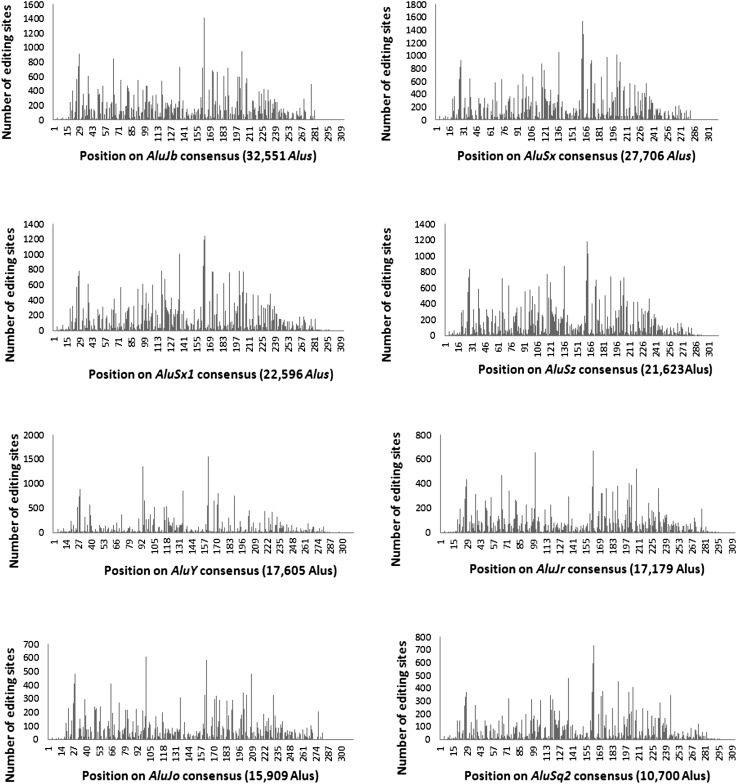
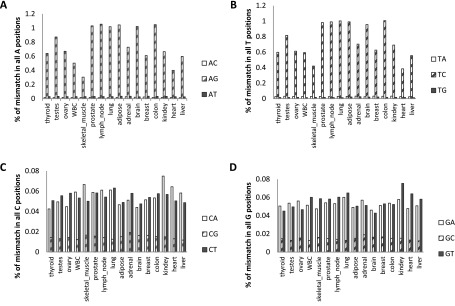
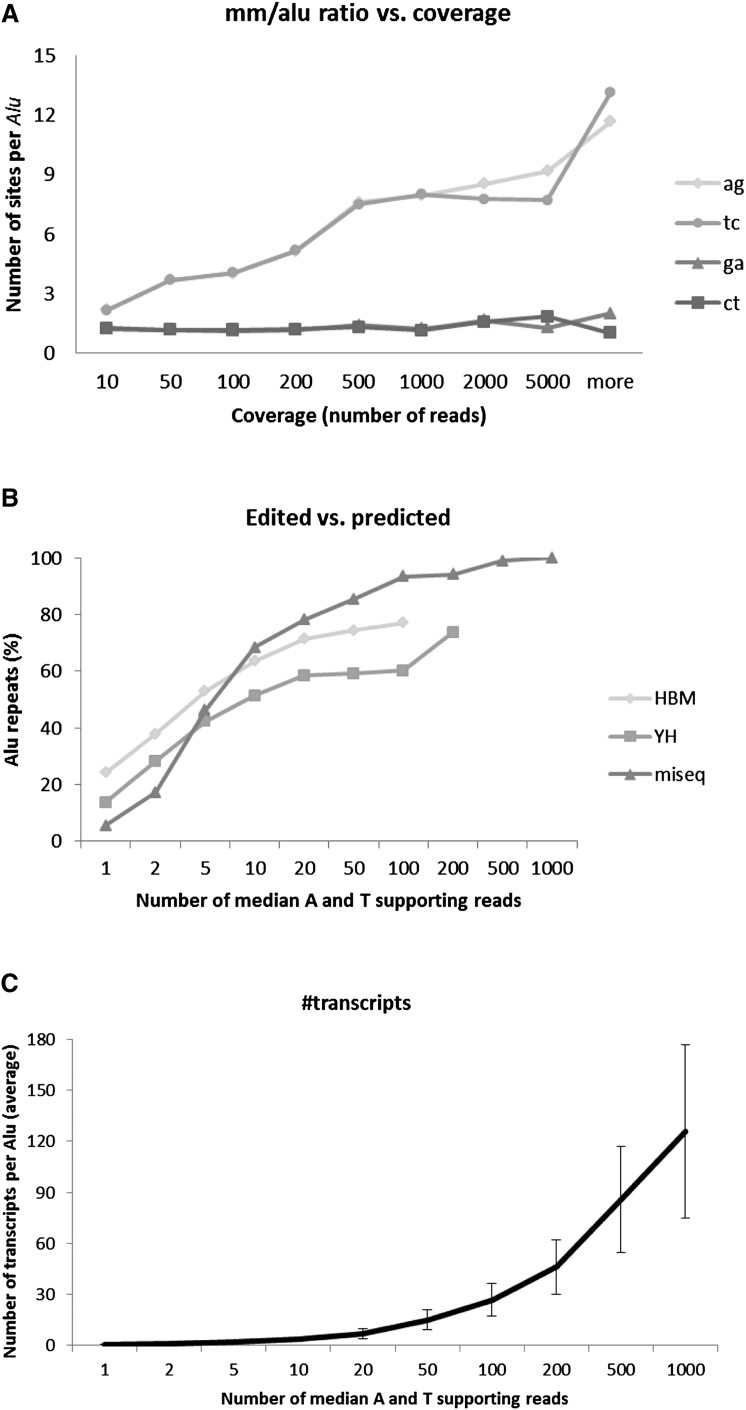
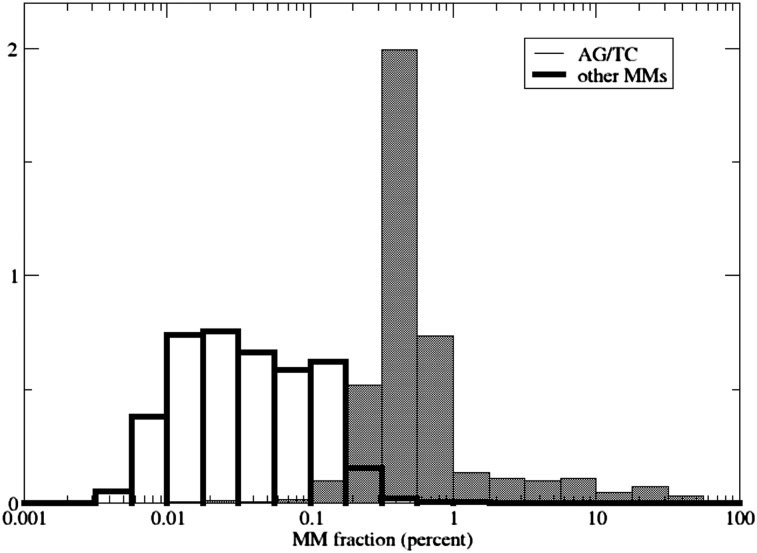
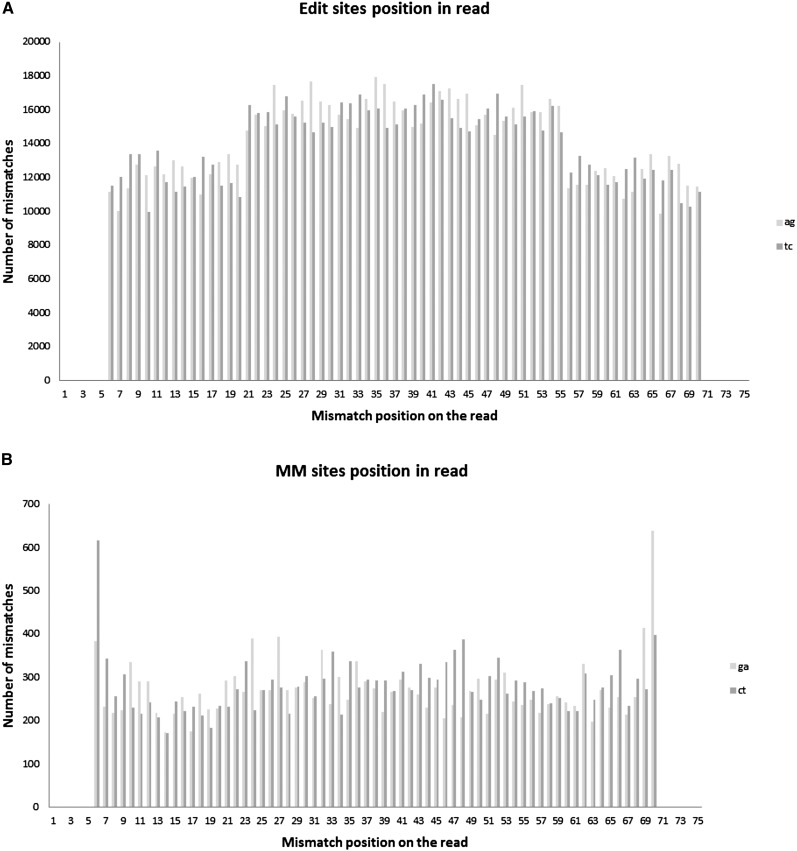
RNA molecules transmit the information encoded in the genome and generally reflect its content. Adenosine-to-inosine (A-to-I) RNA editing by ADAR proteins converts a genomically encoded adenosine into inosine. It is known that most RNA editing in human takes place in the primate-specific Alu sequences, but the extent of this phenomenon and its effect on transcriptome diversity are not yet clear. Here, we analyzed large-scale RNA-seq data and detected ∼1.6 million editing sites. As detection sensitivity increases with sequencing coverage, we performed ultradeep sequencing of selected Alu sequences and showed that the scope of editing is much larger than anticipated. We found that virtually all adenosines within Alu repeats that form double-stranded RNA undergo A-to-I editing, although most sites exhibit editing at only low levels (<1%). Moreover, using high coverage sequencing, we observed editing of transcripts resulting from residual antisense expression, doubling the number of edited sites in the human genome. Based on bioinformatic analyses and deep targeted sequencing, we estimate that there are over 100 million human Alu RNA editing sites, located in the majority of human genes. These findings set the stage for exploring how this primate-specific massive diversification of the transcriptome is utilized.
Figures
Similar articles
-
Alu elements shape the primate transcriptome by cis-regulation of RNA editing.Genome Biol. 2014 Feb 3;15(2):R28. doi: 10.1186/gb-2014-15-2-r28. Genome Biol. 2014. PMID: 24485196 Free PMC article.
-
One hundred million adenosine-to-inosine RNA editing sites: hearing through the noise.Bioessays. 2014 Aug;36(8):730-5. doi: 10.1002/bies.201400055. Epub 2014 May 30. Bioessays. 2014. PMID: 24889193 Free PMC article.
-
ALU A-to-I RNA Editing: Millions of Sites and Many Open Questions.Methods Mol Biol. 2021;2181:149-162. doi: 10.1007/978-1-0716-0787-9_9. Methods Mol Biol. 2021. PMID: 32729079 Review.
-
Letter from the editor: Adenosine-to-inosine RNA editing in Alu repeats in the human genome.EMBO Rep. 2005 Sep;6(9):831-5. doi: 10.1038/sj.embor.7400507. EMBO Rep. 2005. PMID: 16138094 Free PMC article. Review.
-
Identification of widespread ultra-edited human RNAs.PLoS Genet. 2011 Oct;7(10):e1002317. doi: 10.1371/journal.pgen.1002317. Epub 2011 Oct 20. PLoS Genet. 2011. PMID: 22028664 Free PMC article.
Cited by
-
The Regulation of RNA Modification Systems: The Next Frontier in Epitranscriptomics?Genes (Basel). 2021 Feb 26;12(3):345. doi: 10.3390/genes12030345. Genes (Basel). 2021. PMID: 33652758 Free PMC article. Review.
-
Detecting haplotype-specific transcript variation in long reads with FLAIR2.Genome Biol. 2024 Jul 2;25(1):173. doi: 10.1186/s13059-024-03301-y. Genome Biol. 2024. PMID: 38956576 Free PMC article.
-
MiRNA post-transcriptional modification dynamics in T cell activation.iScience. 2021 May 12;24(6):102530. doi: 10.1016/j.isci.2021.102530. eCollection 2021 Jun 25. iScience. 2021. PMID: 34142042 Free PMC article.
-
Recent Advances in Adenosine-to-Inosine RNA Editing in Cancer.Cancer Treat Res. 2023;190:143-179. doi: 10.1007/978-3-031-45654-1_5. Cancer Treat Res. 2023. PMID: 38113001 Review.
-
RNA epitranscriptomics dysregulation: A major determinant for significantly increased risk of ASD pathogenesis.Front Neurosci. 2023 Feb 16;17:1101422. doi: 10.3389/fnins.2023.1101422. eCollection 2023. Front Neurosci. 2023. PMID: 36875672 Free PMC article.
References
-
- Bass BL, Weintraub H 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials