

Assessment of ligand binding site predictions in CASP10
- PMID: 24339001
- PMCID: PMC4495912
- DOI: 10.1002/prot.24495
Assessment of ligand binding site predictions in CASP10
Abstract
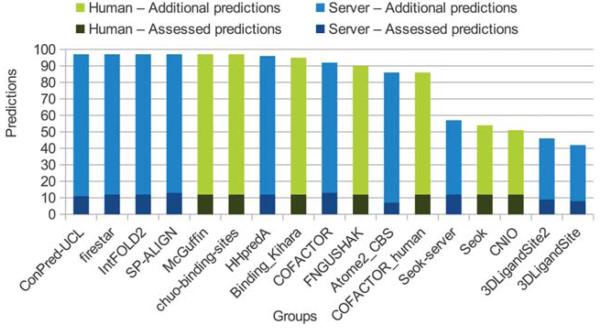
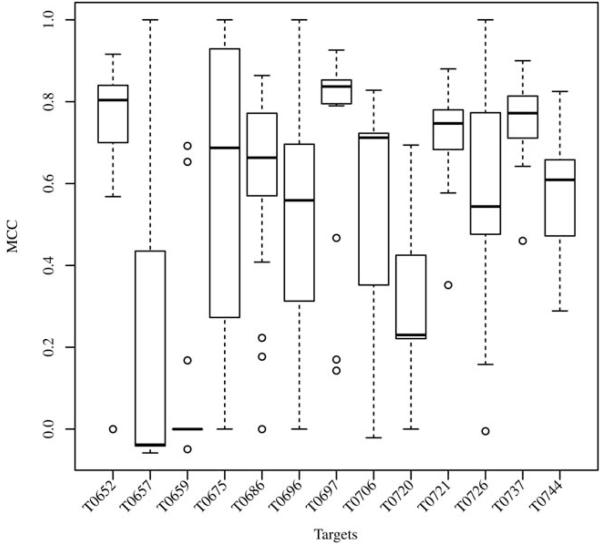
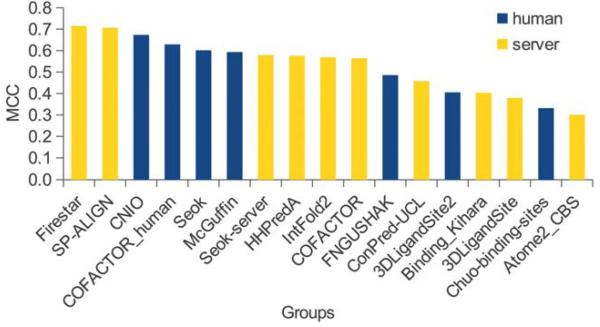
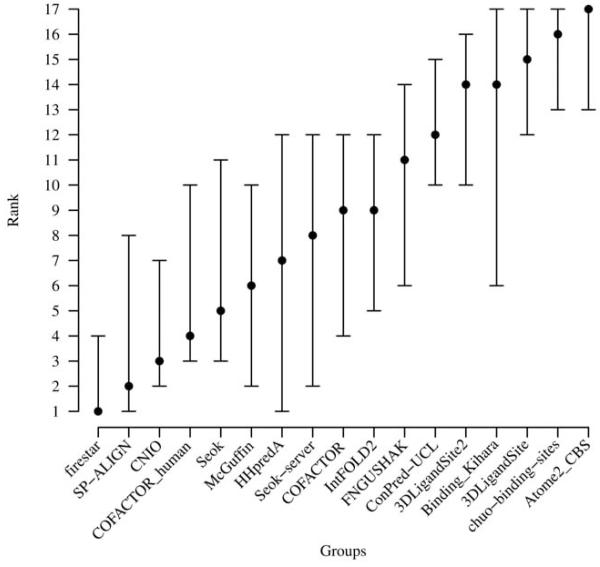
The identification of amino acid residues in proteins involved in binding small molecule ligands is an important step for their functional characterization, as the function of a protein often depends on specific interactions with other molecules. The accuracy of computational methods aiming to predict such binding residues was evaluated within the "function prediction (prediction of binding sites, FN)" category of the critical assessment of protein structure prediction (CASP) experiment. In the last edition of the experiment (CASP10), 17 research groups participated in this category, and their predictions were evaluated on 13 prediction targets containing biologically relevant ligands. The results of this experiment indicate that several methods achieved an overall good performance, showing the usefulness of such methods in predicting ligand binding residues. As in previous years, methods based on a homology transfer approach were dominating. In comparison to CASP9, a larger fraction of the top predictors are automated servers. However, due to the small number of targets and the characteristics of the prediction format, the differences observed among the first ten methods were not statistically significant and it was also not possible to analyze differences in accuracy for different ligand types or overall structure, difficulty. To overcome these limitations and to allow for a more detailed evaluation, in future editions of CASP, methods in the FN category will no longer be evaluated on the "normal" CASP targets, but assessed continuously by CAMEO (continuous automated model evaluation) based on weekly prereleased sequences from the PDB.
Keywords: CASP; active site; assessment; binding site; cofactor; evaluation; ligand; protein function; protein structure.
Copyright © 2013 Wiley Periodicals, Inc.
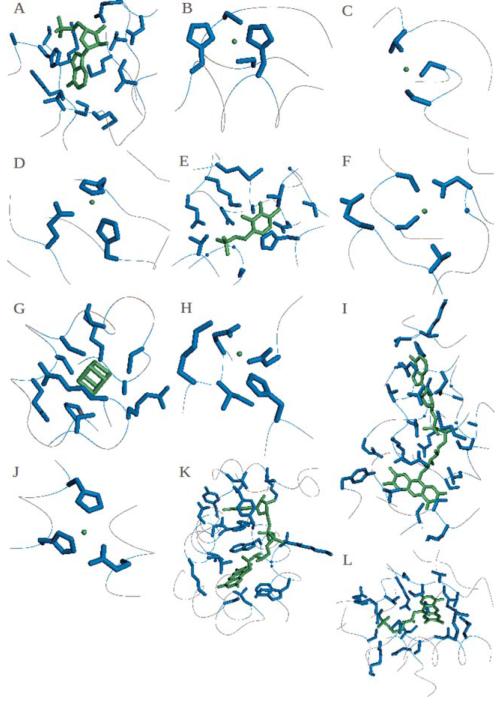
Figures
Similar articles
-
Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12.Proteins. 2018 Mar;86 Suppl 1(Suppl 1):387-398. doi: 10.1002/prot.25431. Epub 2017 Dec 17. Proteins. 2018. PMID: 29178137 Free PMC article.
-
Introducing "best single template" models as reference baseline for the Continuous Automated Model Evaluation (CAMEO).Proteins. 2019 Dec;87(12):1378-1387. doi: 10.1002/prot.25815. Epub 2019 Oct 16. Proteins. 2019. PMID: 31571280 Free PMC article.
-
Assessment of ligand-binding residue predictions in CASP9.Proteins. 2011;79 Suppl 10(Suppl 10):126-36. doi: 10.1002/prot.23174. Epub 2011 Oct 11. Proteins. 2011. PMID: 21987472 Free PMC article.
-
Review and comparative assessment of sequence-based predictors of protein-binding residues.Brief Bioinform. 2018 Sep 28;19(5):821-837. doi: 10.1093/bib/bbx022. Brief Bioinform. 2018. PMID: 28334258 Review.
-
Protein structure prediction: challenging targets for CASP10.J Biomol Struct Dyn. 2012;30(5):607-15. doi: 10.1080/07391102.2012.687526. Epub 2012 Jun 26. J Biomol Struct Dyn. 2012. PMID: 22731875 Review.
Cited by
-
Biological function derived from predicted structures in CASP11.Proteins. 2016 Sep;84 Suppl 1(Suppl 1):370-91. doi: 10.1002/prot.24997. Epub 2016 Jun 15. Proteins. 2016. PMID: 27181425 Free PMC article.
-
Critical assessment of methods of protein structure prediction (CASP)--round x.Proteins. 2014 Feb;82 Suppl 2(0 2):1-6. doi: 10.1002/prot.24452. Epub 2013 Dec 17. Proteins. 2014. PMID: 24344053 Free PMC article.
-
New prediction categories in CASP15.Proteins. 2023 Dec;91(12):1550-1557. doi: 10.1002/prot.26515. Epub 2023 Jun 12. Proteins. 2023. PMID: 37306011 Free PMC article.
-
IntFOLD: an integrated server for modelling protein structures and functions from amino acid sequences.Nucleic Acids Res. 2015 Jul 1;43(W1):W169-73. doi: 10.1093/nar/gkv236. Epub 2015 Mar 27. Nucleic Acids Res. 2015. PMID: 25820431 Free PMC article.
-
A multilayer dynamic perturbation analysis method for predicting ligand-protein interactions.BMC Bioinformatics. 2022 Nov 2;23(1):456. doi: 10.1186/s12859-022-04995-2. BMC Bioinformatics. 2022. PMID: 36324073 Free PMC article.
References
-
- Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N. Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics. 2002;18(Suppl 1):S71–77. - PubMed
-
- Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23(15):1875–1882. - PubMed
-
- Fischer JD, Mayer CE, Soding J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 2008;24(5):613–620. - PubMed
-
- Laurie AT, Jackson RM. Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites. Bioinformatics. 2005;21(9):1908–1916. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
