

Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS
- PMID: 24184128
- PMCID: PMC7113851
- DOI: 10.1016/j.antiviral.2013.10.013
Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS
Abstract
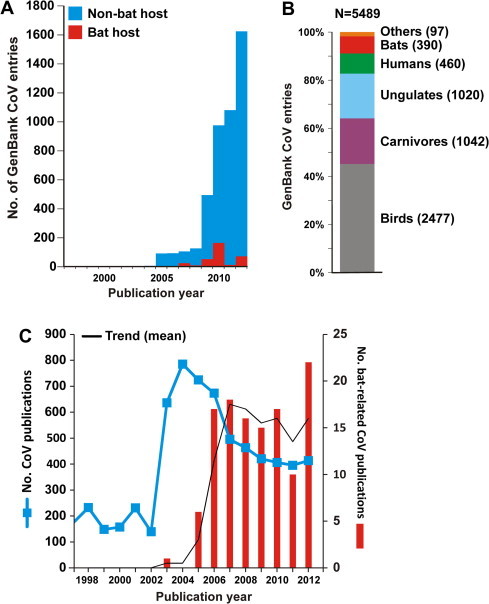
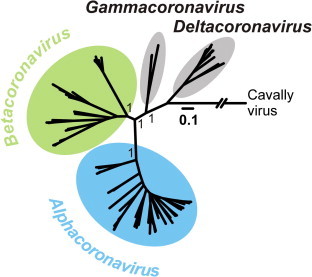
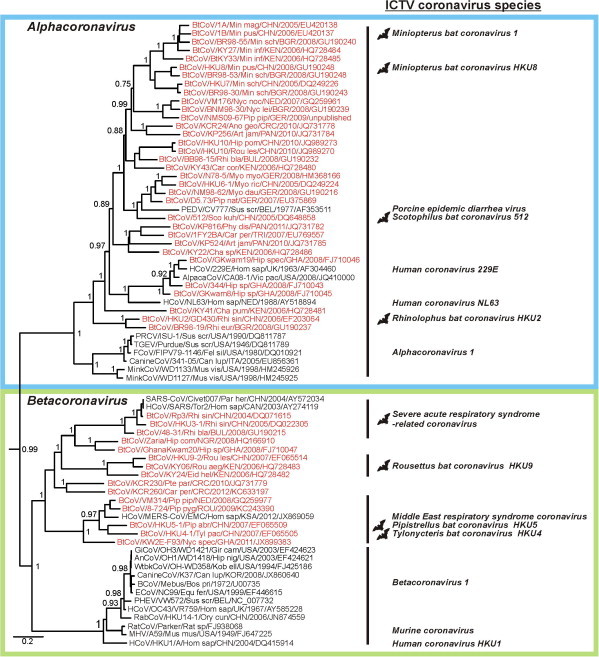
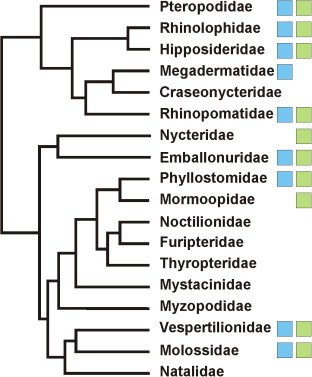
In 2002/2003, a novel coronavirus (CoV) caused a pandemic, infecting more than 8000 people, of whom nearly 10% died. This virus, termed severe acute respiratory syndrome-CoV was linked to a zoonotic origin from rhinolophid bats in 2005. Since then, numerous studies have described novel bat CoVs, including close relatives of the newly emerging Middle East respiratory syndrome (MERS)-CoV. In this paper we discuss CoV genomic properties and compare different taxonomic approaches in light of the technical difficulties of obtaining full genomic sequences directly from bat specimens. We first present an overview of the available studies on bat CoVs, with details on their chiropteran hosts, then comparatively analyze the increase in bat CoV studies and novel genomic sequences obtained since the SARS pandemic. We then conduct a comprehensive phylogenetic analysis of the genera Alpha- and Betacoronavirus, to show that bats harbour more CoV diversity than other mammalian hosts and are widely represented in most, but not all parts of the tree of mammalian CoVs. We next discuss preliminary evidence for phylogenetic co-segregation of CoVs and bat hosts encompassing the Betacoronavirus clades b and d, with an emphasis on the sampling bias that exists among bat species and other mammals, then present examples of CoVs infecting different hosts on the one hand and viruses apparently confined to host genera on the other. We also demonstrate a geographic bias within available studies on bat CoVs, and identify a critical lack of information from biodiversity hotspots in Africa, Asia and Latin America. We then present evidence for a zoonotic origin of four of the six known human CoVs (HCoV), three of which likely involved bats, namely SARS-CoV, MERS-CoV and HCoV-229E; compare the available data on CoV pathogenesis in bats to that in other mammalian hosts; and discuss hypotheses on the putative insect origins of CoV ancestors. Finally, we suggest caution with conclusions on the zoonotic potential of bat viruses, based only on genomic sequence data, and emphasize the need to preserve these ecologically highly relevant animals. This paper forms part of a symposium in Antiviral Research on "from SARS to MERS: 10years of research on highly pathogenic human coronaviruses".
Keywords: Alphacoronavirus; Bats; Betacoronavirus; Coronaviridae; Taxonomy; Zoonosis.
Copyright © 2013 Elsevier B.V. All rights reserved.
Figures
Similar articles
-
Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History.J Virol. 2017 Feb 14;91(5):e01953-16. doi: 10.1128/JVI.01953-16. Print 2017 Mar 1. J Virol. 2017. PMID: 28077633 Free PMC article.
-
[Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus).].Vopr Virusol. 2020;65(2):62-70. doi: 10.36233/0507-4088-2020-65-2-62-70. Vopr Virusol. 2020. PMID: 32515561 Review. Russian.
-
Evidence for an Ancestral Association of Human Coronavirus 229E with Bats.J Virol. 2015 Dec;89(23):11858-70. doi: 10.1128/JVI.01755-15. Epub 2015 Sep 16. J Virol. 2015. PMID: 26378164 Free PMC article.
-
Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China.J Virol. 2014 Jun;88(12):7070-82. doi: 10.1128/JVI.00631-14. Epub 2014 Apr 9. J Virol. 2014. PMID: 24719429 Free PMC article.
-
Bat origin of human coronaviruses.Virol J. 2015 Dec 22;12:221. doi: 10.1186/s12985-015-0422-1. Virol J. 2015. PMID: 26689940 Free PMC article. Review.
Cited by
-
MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels.Virus Res. 2015 Apr 2;201:61-6. doi: 10.1016/j.virusres.2015.02.023. Epub 2015 Feb 27. Virus Res. 2015. PMID: 25733052 Free PMC article.
-
Origin and cross-species transmission of bat coronaviruses in China.Nat Commun. 2020 Aug 25;11(1):4235. doi: 10.1038/s41467-020-17687-3. Nat Commun. 2020. Retraction in: Nat Commun. 2024 Dec 19;15(1):10706. doi: 10.1038/s41467-024-55454-w PMID: 32843626 Free PMC article. Retracted.
-
The Safety of Slaughterhouse Workers during the Pandemic Crisis.Int J Environ Res Public Health. 2021 Mar 5;18(5):2633. doi: 10.3390/ijerph18052633. Int J Environ Res Public Health. 2021. PMID: 33807936 Free PMC article.
-
Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations.Viruses. 2021 May 18;13(5):936. doi: 10.3390/v13050936. Viruses. 2021. PMID: 34070175 Free PMC article. Review.
-
Potential zoonotic sources of SARS-CoV-2 infections.Transbound Emerg Dis. 2021 Jul;68(4):1824-1834. doi: 10.1111/tbed.13872. Epub 2020 Oct 23. Transbound Emerg Dis. 2021. PMID: 33034151 Free PMC article. Review.
References
-
- Alekseev K.P., Vlasova A.N., Jung K., Hasoksuz M., Zhang X., Halpin R., Wang S., Ghedin E., Spiro D., Saif L.J. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. J. Virol. 2008;82:12422–12431. - PMC - PubMed
-
- Amman B.R., Carroll S.A., Reed Z.D., Sealy T.K., Balinandi S., Swanepoel R., Kemp A., Erickson B.R., Comer J.A., Campbell S., Cannon D.L., Khristova M.L., Atimnedi P., Paddock C.D., Crockett R.J., Flietstra T.D., Warfield K.L., Unfer R., Katongole-Mbidde E., Downing R., Tappero J.W., Zaki S.R., Rollin P.E., Ksiazek T.G., Nichol S.T., Towner J.S. Seasonal pulses of Marburg virus circulation in juvenile Rousettus aegyptiacus bats coincide with periods of increased risk of human infection. PLoS Pathog. 2012;8:e1002877. - PMC - PubMed
-
- Annan A., Baldwin H.J., Corman V.M., Klose S.M., Owusu M., Nkrumah E.E., Badu E.K., Anti P., Agbenyega O., Meyer B., Oppong S., Sarkodie Y.A., Kalko E.K., Lina P.H., Godlevska E.V., Reusken C., Seebens A., Gloza-Rausch F., Vallo P., Tschapka M., Drosten C., Drexler J.F. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013;19:456–459. - PMC - PubMed
-
- Anthony S., Ojeda-Flores R., Rico-Chavez O., Navarrete-Macias I., Zambrana-Torrelio C., Rostal M.K., Epstein J.H., Tipps T., Liang E., Sanchez-Leon M., Sotomayor-Bonilla J., Aguirre A.A., Avila R., Medellin R.A., Goldstein T., Suzan G., Daszak P., Lipkin W.I. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013 - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
