

Inference of the properties of the recombination process from whole bacterial genomes
- PMID: 24172133
- PMCID: PMC3872189
- DOI: 10.1534/genetics.113.157172
Inference of the properties of the recombination process from whole bacterial genomes
Abstract
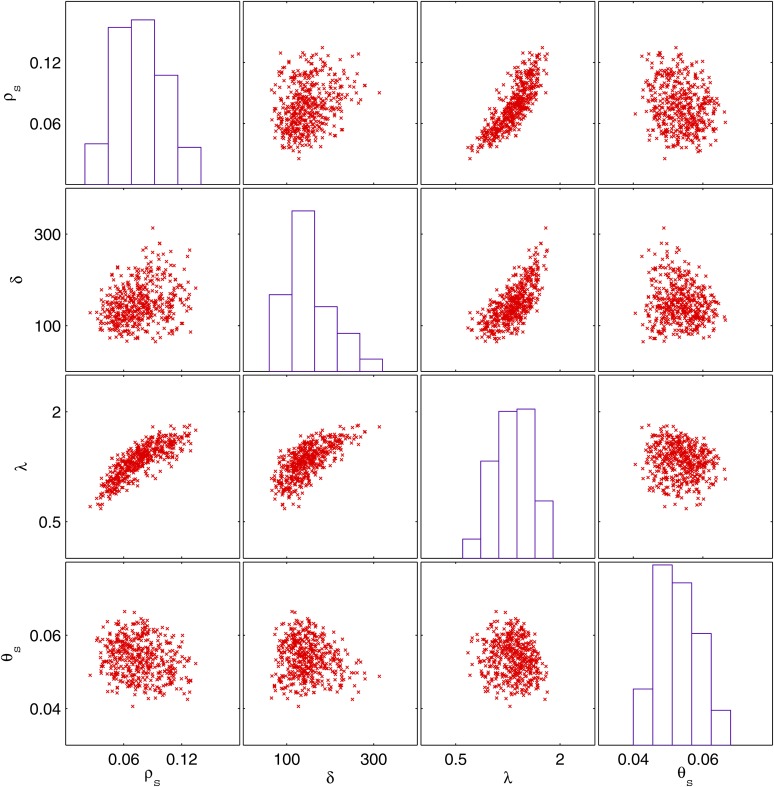
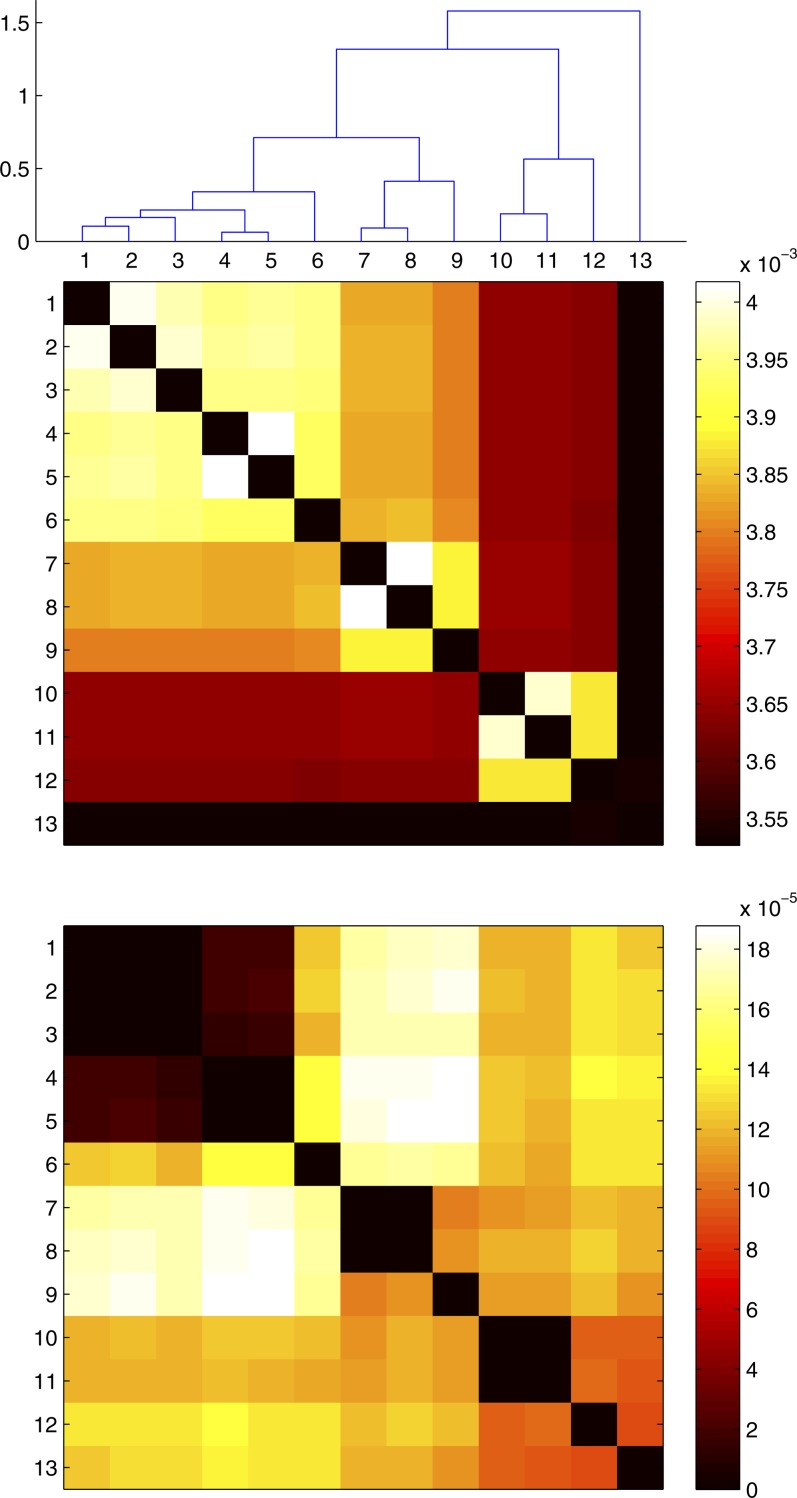
Patterns of linkage disequilibrium, homoplasy, and incompatibility are difficult to interpret because they depend on several factors, including the recombination process and the population structure. Here we introduce a novel model-based framework to infer recombination properties from such summary statistics in bacterial genomes. The underlying model is sequentially Markovian so that data can be simulated very efficiently, and we use approximate Bayesian computation techniques to infer parameters. As this does not require us to calculate the likelihood function, the model can be easily extended to investigate less probed aspects of recombination. In particular, we extend our model to account for the bias in the recombination process whereby closely related bacteria recombine more often with one another. We show that this model provides a good fit to a data set of Bacillus cereus genomes and estimate several recombination properties, including the rate of bias in recombination. All the methods described in this article are implemented in a software package that is freely available for download at http://code.google.com/p/clonalorigin/.
Keywords: bacteria; biased recombination; four-gamete test (G4); homologous recombination; homology-dependent recombination; homoplasy; linkage disequilibrium (LD).
Figures
Similar articles
-
Inference of homologous recombination in bacteria using whole-genome sequences.Genetics. 2010 Dec;186(4):1435-49. doi: 10.1534/genetics.110.120121. Epub 2010 Oct 5. Genetics. 2010. PMID: 20923983 Free PMC article.
-
The Bacterial Sequential Markov Coalescent.Genetics. 2017 May;206(1):333-343. doi: 10.1534/genetics.116.198796. Epub 2017 Mar 3. Genetics. 2017. PMID: 28258183 Free PMC article.
-
Recombination hotspots as a point process.Philos Trans R Soc Lond B Biol Sci. 2005 Aug 29;360(1460):1597-603. doi: 10.1098/rstb.2005.1690. Philos Trans R Soc Lond B Biol Sci. 2005. PMID: 16096109 Free PMC article.
-
Ancestral Population Genomics.Methods Mol Biol. 2019;1910:555-589. doi: 10.1007/978-1-4939-9074-0_18. Methods Mol Biol. 2019. PMID: 31278677 Review.
-
Corn and humans: recombination and linkage disequilibrium in two genomes of similar size.Trends Genet. 2004 Feb;20(2):103-11. doi: 10.1016/j.tig.2003.12.002. Trends Genet. 2004. PMID: 14746992 Review.
Cited by
-
Coalescent framework for prokaryotes undergoing interspecific homologous recombination.Heredity (Edinb). 2018 May;120(5):474-484. doi: 10.1038/s41437-017-0034-1. Epub 2018 Jan 23. Heredity (Edinb). 2018. PMID: 29358726 Free PMC article.
-
Rates of Molecular Evolution in a Marine Synechococcus Phage Lineage.Viruses. 2019 Aug 6;11(8):720. doi: 10.3390/v11080720. Viruses. 2019. PMID: 31390807 Free PMC article.
-
Correlated Mutations and Homologous Recombination Within Bacterial Populations.Genetics. 2017 Feb;205(2):891-917. doi: 10.1534/genetics.116.189621. Epub 2016 Dec 22. Genetics. 2017. PMID: 28007887 Free PMC article.
-
Core genes can have higher recombination rates than accessory genes within global microbial populations.Elife. 2022 Jul 8;11:e78533. doi: 10.7554/eLife.78533. Elife. 2022. PMID: 35801696 Free PMC article.
-
Geographic population structure and distinct intra-population dynamics of globally abundant freshwater bacteria.ISME J. 2024 Jan 8;18(1):wrae113. doi: 10.1093/ismejo/wrae113. ISME J. 2024. PMID: 38959851 Free PMC article.
References
-
- Achtman M., Wagner M., 2008. Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 6: 431–440. - PubMed
-
- Beaumont M. A., 2010. Approximate Bayesian computation in evolution and ecology. Annu. Rev. Ecol. Evol. Syst. 41: 379–406.
-
- Beaumont M. A., Cornuet J. M., Marin J. M., Robert C. P., 2009. Adaptive approximate Bayesian computation. Biometrika 96: 983–990.
-
- Brockwell A. E., 2006. Parallel Markov chain Monte Carlo simulation by pre-fetching. J. Comput. Graph. Stat. 15: 246–261.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
