

Experimental induction of type 2 diabetes in aging-accelerated mice triggered Alzheimer-like pathology and memory deficits
- PMID: 24121970
- PMCID: PMC3941701
- DOI: 10.3233/JAD-131238
Experimental induction of type 2 diabetes in aging-accelerated mice triggered Alzheimer-like pathology and memory deficits
Abstract
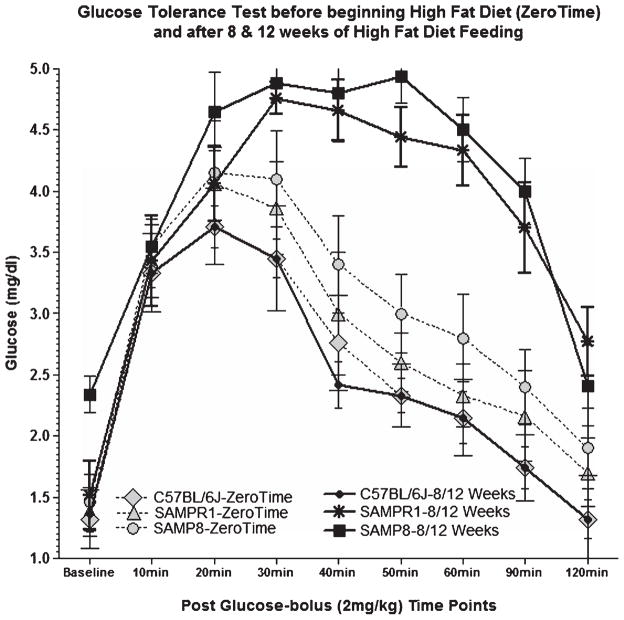
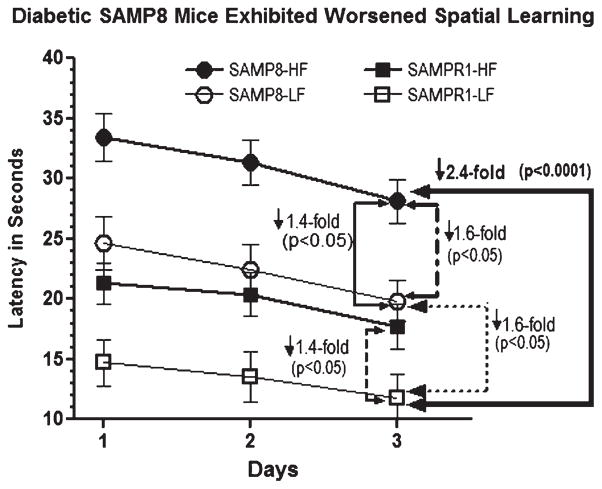
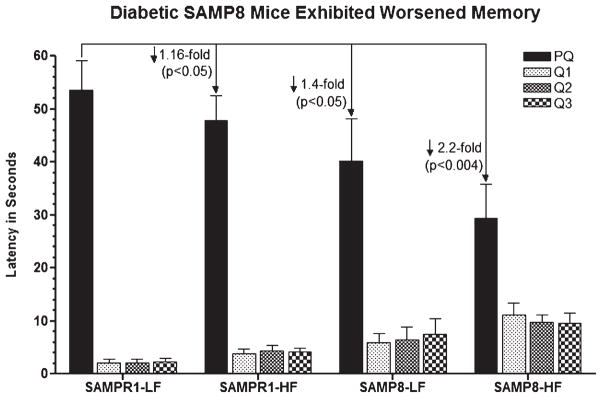
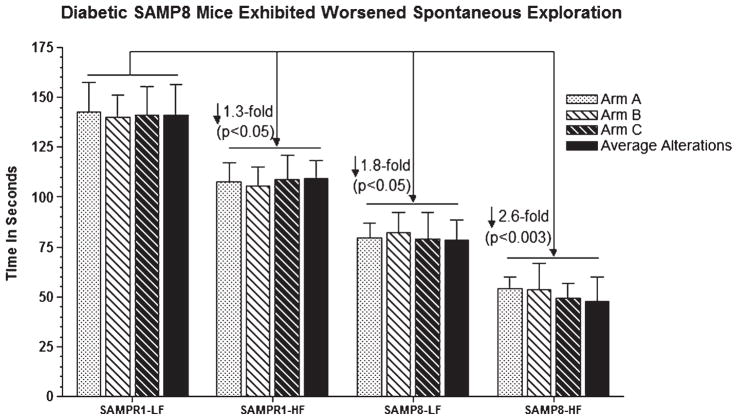
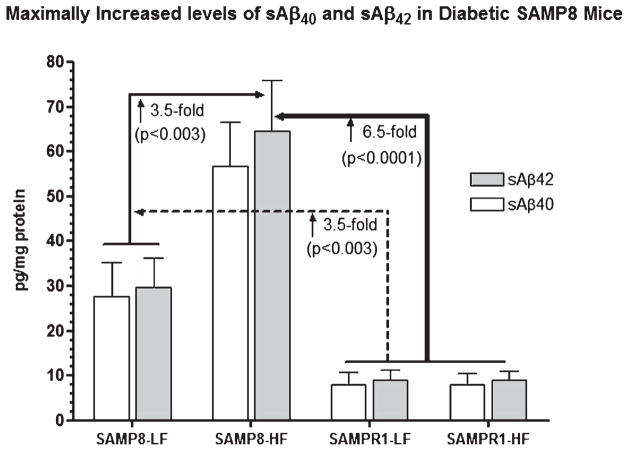
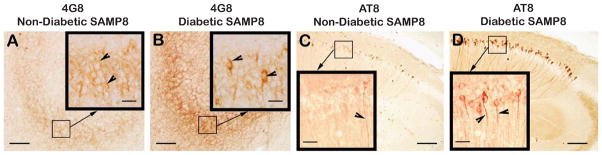
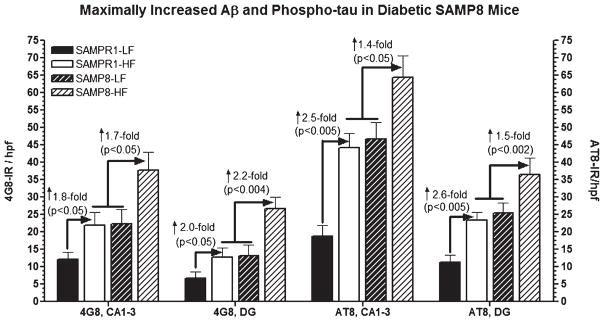
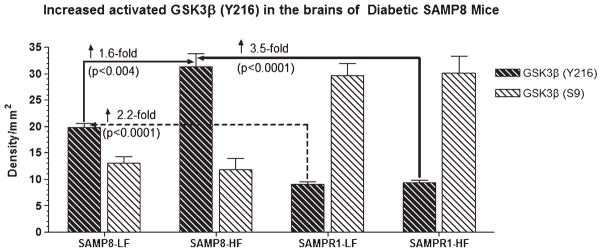
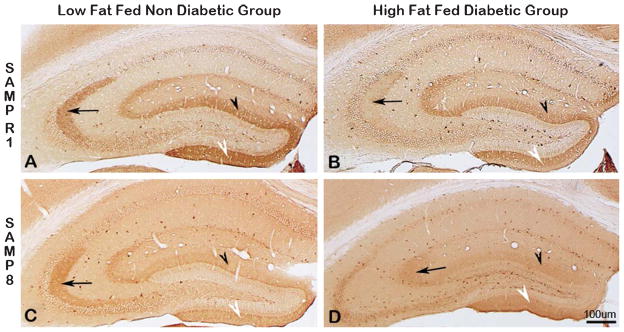
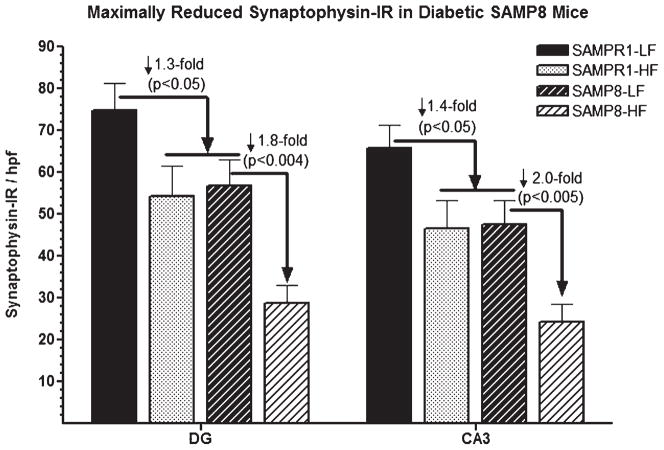
Alzheimer's disease (AD) is an age-dependent neurodegenerative disease constituting ~95% of late-onset non-familial/sporadic AD, and only ~5% accounting for early-onset familial AD. Availability of a pertinent model representing sporadic AD is essential for testing candidate therapies. Emerging evidence indicates a causal link between diabetes and AD. People with diabetes are >1.5-fold more likely to develop AD. Senescence-accelerated mouse model (SAMP8) of accelerated aging displays many features occurring early in AD. Given the role played by diabetes in the pre-disposition of AD, and the utility of SAMP8 non-transgenic mouse model of accelerated aging, we examined if high fat diet-induced experimental type 2 diabetes in SAMP8 mice will trigger pathological aging of the brain. Results showed that compared to non-diabetic SAMP8 mice, diabetic SAMP8 mice exhibited increased cerebral amyloid-β, dysregulated tau-phosphorylating glycogen synthase kinase 3β, reduced synaptophysin immunoreactivity, and displayed memory deficits, indicating Alzheimer-like changes. High fat diet-induced type 2 diabetic SAMP8 mice may represent the metabolic model of AD.
Keywords: Alzheimer's disease; amyloid-β; diabetes; glycogen synthase kinase-3β; learning and memory; pathological aging of the brain; senescence-accelerated mice; synaptophysin; tau.
Figures
Similar articles
-
The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer's disease animal model.Ageing Res Rev. 2014 Jan;13:13-37. doi: 10.1016/j.arr.2013.10.002. Epub 2013 Nov 21. Ageing Res Rev. 2014. PMID: 24269312
-
The GLP-1 Receptor Agonist Liraglutide Improves Memory Function and Increases Hippocampal CA1 Neuronal Numbers in a Senescence-Accelerated Mouse Model of Alzheimer's Disease.J Alzheimers Dis. 2015;46(4):877-88. doi: 10.3233/JAD-143090. J Alzheimers Dis. 2015. PMID: 25869785 Free PMC article.
-
Effects of accelerated senescence on learning and memory, locomotion and anxiety-like behavior in APP/PS1 mouse model of Alzheimer's disease.J Neurol Sci. 2013 Dec 15;335(1-2):145-54. doi: 10.1016/j.jns.2013.09.018. Epub 2013 Sep 21. J Neurol Sci. 2013. PMID: 24095271
-
SAMP8 Mice as a Model of Age-Related Cognition Decline with Underlying Mechanisms in Alzheimer's Disease.J Alzheimers Dis. 2020;75(2):385-395. doi: 10.3233/JAD-200063. J Alzheimers Dis. 2020. PMID: 32310176 Review.
-
From aging to Alzheimer's disease: unveiling "the switch" with the senescence-accelerated mouse model (SAMP8).J Alzheimers Dis. 2008 Dec;15(4):615-24. doi: 10.3233/jad-2008-15408. J Alzheimers Dis. 2008. PMID: 19096160 Review.
Cited by
-
The Glymphatic System in Diabetes-Induced Dementia.Front Neurol. 2018 Oct 31;9:867. doi: 10.3389/fneur.2018.00867. eCollection 2018. Front Neurol. 2018. PMID: 30429819 Free PMC article. Review.
-
Resveratrol Protects SAMP8 Brain Under Metabolic Stress: Focus on Mitochondrial Function and Wnt Pathway.Mol Neurobiol. 2017 Apr;54(3):1661-1676. doi: 10.1007/s12035-016-9770-0. Epub 2016 Feb 12. Mol Neurobiol. 2017. PMID: 26873850
-
Fingolimod Modulates the Gene Expression of Proteins Engaged in Inflammation and Amyloid-Beta Metabolism and Improves Exploratory and Anxiety-Like Behavior in Obese Mice.Neurotherapeutics. 2023 Sep;20(5):1388-1404. doi: 10.1007/s13311-023-01403-2. Epub 2023 Jul 11. Neurotherapeutics. 2023. PMID: 37432552 Free PMC article.
-
Reduced neurotrophic factor level is the early event before the functional neuronal deficiency in high-fat diet induced obese mice.Metab Brain Dis. 2017 Feb;32(1):247-257. doi: 10.1007/s11011-016-9905-z. Epub 2016 Sep 13. Metab Brain Dis. 2017. PMID: 27624843
-
Diabetes mellitus accelerates Aβ pathology in brain accompanied by enhanced GAβ generation in nonhuman primates.PLoS One. 2015 Feb 12;10(2):e0117362. doi: 10.1371/journal.pone.0117362. eCollection 2015. PLoS One. 2015. PMID: 25675436 Free PMC article.
References
-
- Finder VH. Alzheimer’s disease: A general introduction and pathomechanism. J Alzheimers Dis. 2010;22(Suppl 3):5–19. - PubMed
-
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. - PubMed
-
- Bastos Leite AJ, Scheltens P, Barkhof F. Pathological aging of the brain: An overview. Top Magn Reson Imaging. 2004;15:369–389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
