

An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis
- PMID: 24120361
- PMCID: PMC3873857
- DOI: 10.1016/j.immuni.2013.09.002
An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis
Abstract
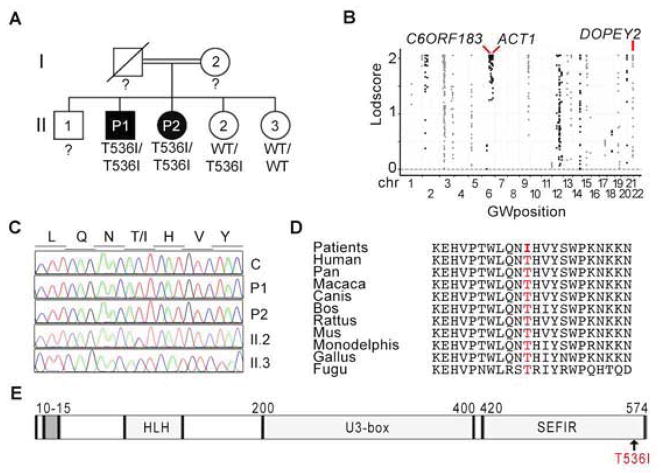
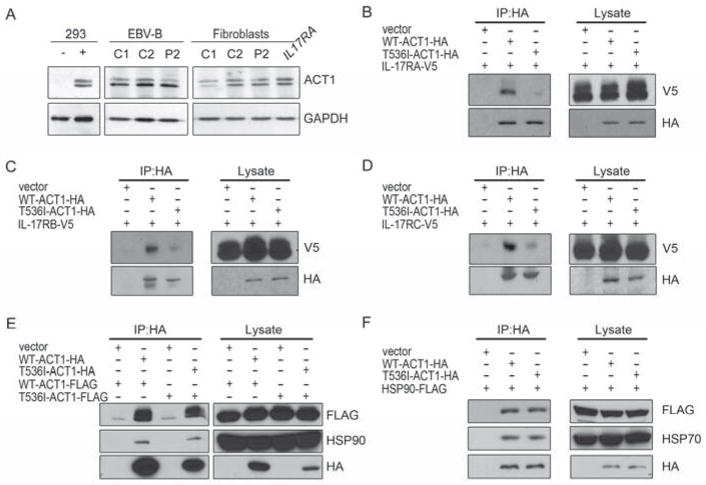
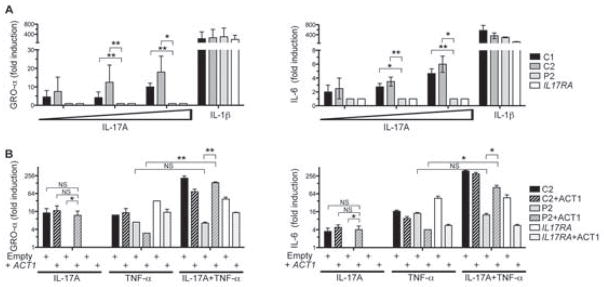
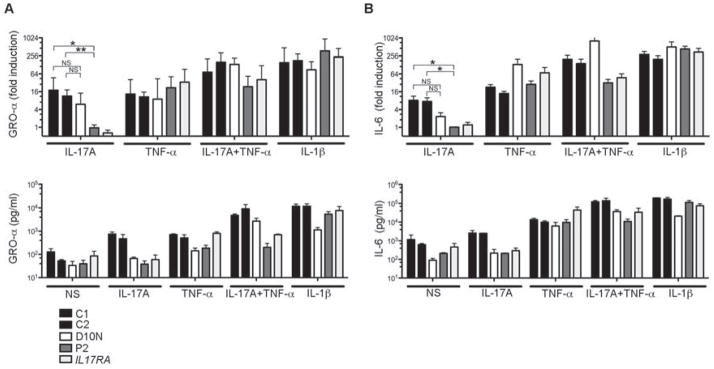
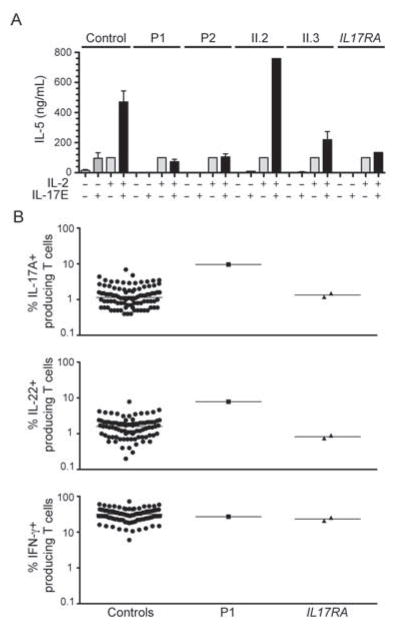
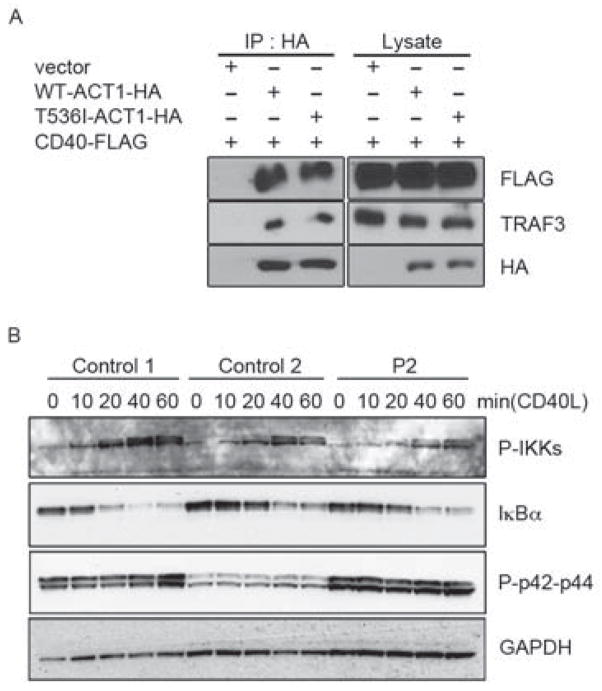
Patients with inborn errors of interleukin-17F (IL-17F) or IL-17RA display chronic mucocutaneous candidiasis (CMC). We report a biallelic missense mutation (T536I) in the adaptor molecule ACT1 in two siblings with CMC. The mutation, located in the SEFIR domain, abolished the homotypic interaction of ACT1 with IL-17 receptors, with no effect on homodimerization. The patients' fibroblasts failed to respond to IL-17A and IL-17F, and their T cells to IL-17E. By contrast, healthy individuals homozygous for the common variant D10N, located in the ACT1 tumor necrosis factor receptor-associated factor-interacting domain and previously associated with psoriasis, had impaired, but not abolished, responses to IL-17 cytokines. SEFIR-independent interactions of ACT1 with other proteins, such as CD40, heat shock protein 70 (HSP70) and HSP90, were not affected by the T536I mutation. Overall, human IL-17A and IL-17F depend on ACT1 to mediate protective mucocutaneous immunity. Moreover, other ACT1-dependent IL-17 cytokines seem to be largely redundant in host defense.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis.J Exp Med. 2015 May 4;212(5):619-31. doi: 10.1084/jem.20141065. Epub 2015 Apr 27. J Exp Med. 2015. PMID: 25918342 Free PMC article.
-
Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity.Science. 2011 Apr 1;332(6025):65-8. doi: 10.1126/science.1200439. Epub 2011 Feb 24. Science. 2011. PMID: 21350122 Free PMC article.
-
The differential regulation of human ACT1 isoforms by Hsp90 in IL-17 signaling.J Immunol. 2014 Aug 15;193(4):1590-9. doi: 10.4049/jimmunol.1400715. Epub 2014 Jul 14. J Immunol. 2014. PMID: 25024377 Free PMC article.
-
Biallelic TRAF3IP2 variants causing chronic mucocutaneous candidiasis in a child harboring a STAT1 variant.Pediatr Allergy Immunol. 2021 Nov;32(8):1804-1812. doi: 10.1111/pai.13603. Epub 2021 Aug 9. Pediatr Allergy Immunol. 2021. PMID: 34289170 Review.
-
Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis.Curr Opin Allergy Clin Immunol. 2012 Dec;12(6):616-22. doi: 10.1097/ACI.0b013e328358cc0b. Curr Opin Allergy Clin Immunol. 2012. PMID: 23026768 Free PMC article. Review.
Cited by
-
Retrospective identification of the first cord blood-transplanted severe aplastic anemia in a STAT1-associated chronic mucocutaneous candidiasis family: case report, review of literature and pathophysiologic background.Front Immunol. 2024 Jul 24;15:1430938. doi: 10.3389/fimmu.2024.1430938. eCollection 2024. Front Immunol. 2024. PMID: 39114664 Free PMC article. Review.
-
Integrated Analysis of Transcriptome Profiles and lncRNA-miRNA-mRNA Competing Endogenous RNA Regulatory Network to Identify Biological Functional Effects of Genes and Pathways Associated with Johne's Disease in Dairy Cattle.Noncoding RNA. 2024 Jun 28;10(4):38. doi: 10.3390/ncrna10040038. Noncoding RNA. 2024. PMID: 39051372 Free PMC article.
-
Candida albicans and Candida glabrata: global priority pathogens.Microbiol Mol Biol Rev. 2024 Jun 27;88(2):e0002123. doi: 10.1128/mmbr.00021-23. Epub 2024 Jun 4. Microbiol Mol Biol Rev. 2024. PMID: 38832801 Review.
-
Friend or Foe - Tc17 cell generation and current evidence for their importance in human disease.Discov Immunol. 2023 Jul 20;2(1):kyad010. doi: 10.1093/discim/kyad010. eCollection 2023. Discov Immunol. 2023. PMID: 38567057 Free PMC article. Review.
-
Isolated Chronic Mucocutaneous Candidiasis due to a Novel Duplication Variant of IL17RC.J Clin Immunol. 2023 Dec 22;44(1):18. doi: 10.1007/s10875-023-01601-9. J Clin Immunol. 2023. PMID: 38129603 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
