

Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance
- PMID: 23979165
- PMCID: PMC4381280
- DOI: 10.1172/JCI67399
Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance
Abstract
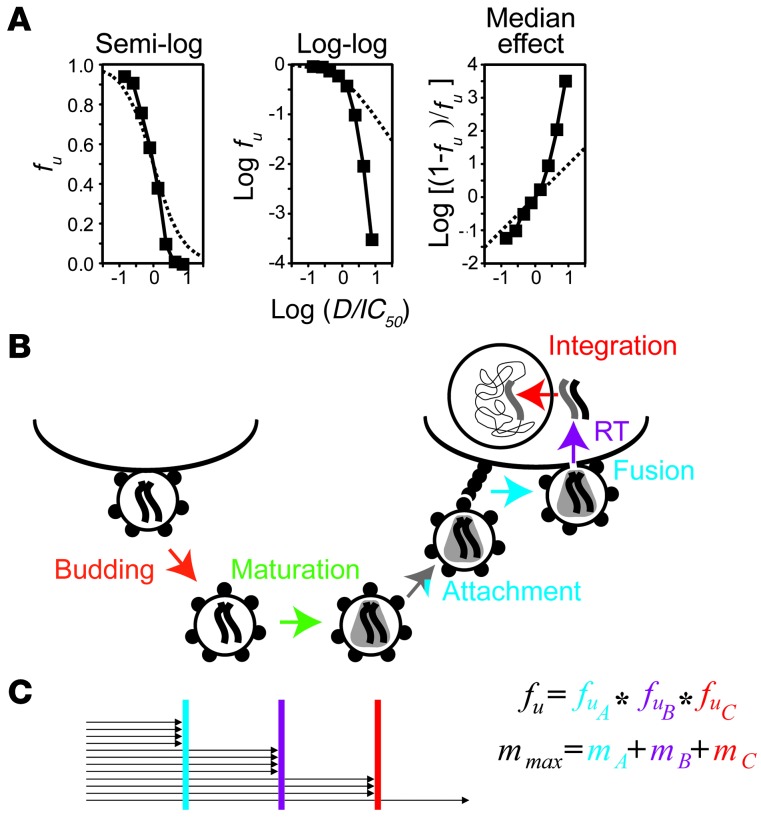
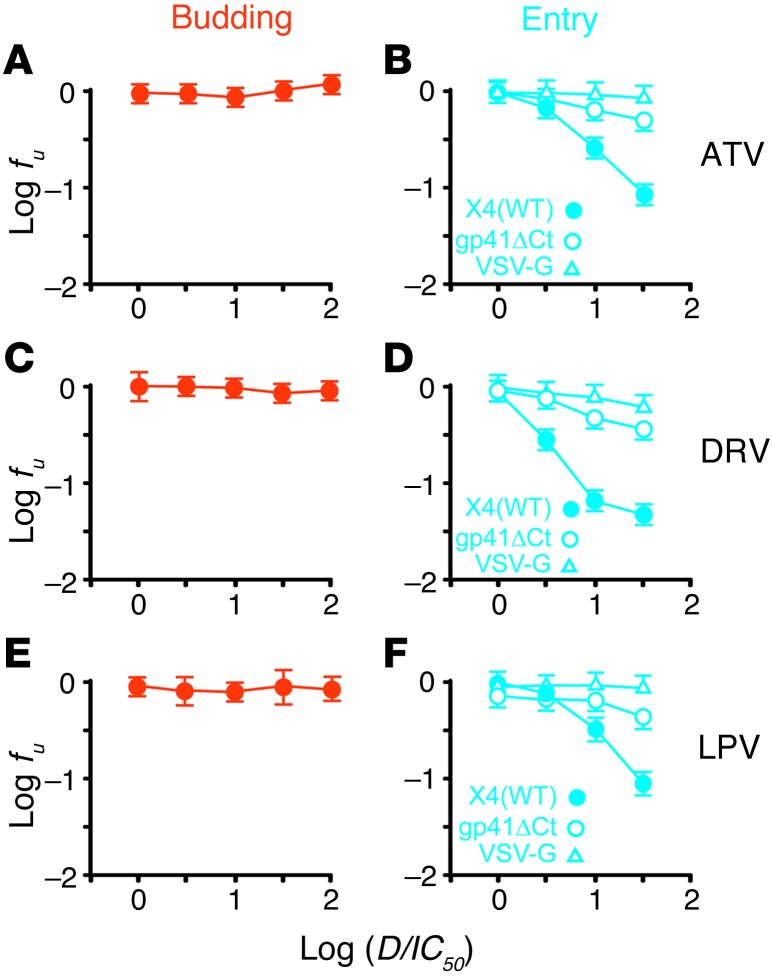
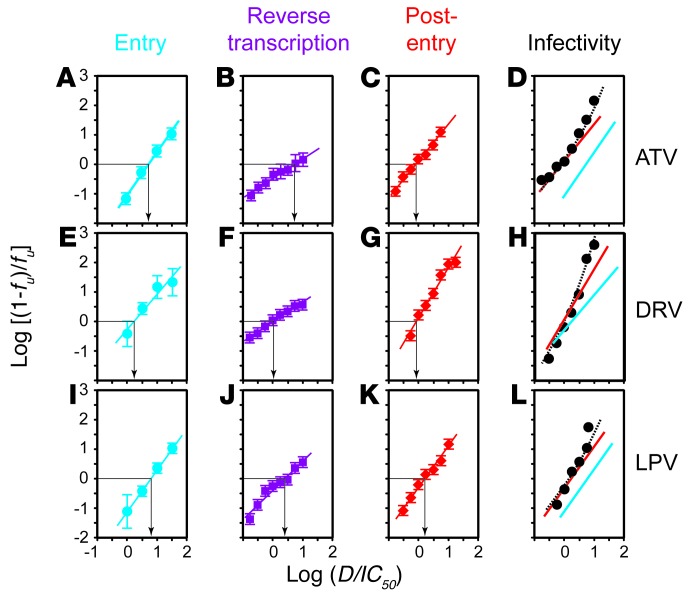
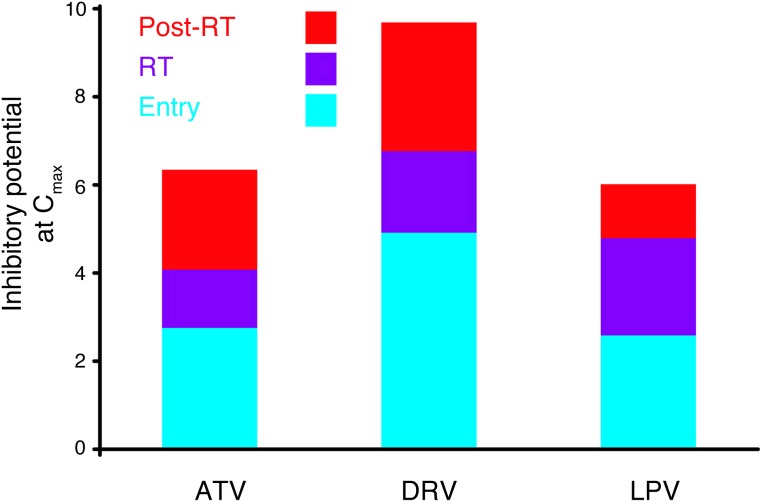
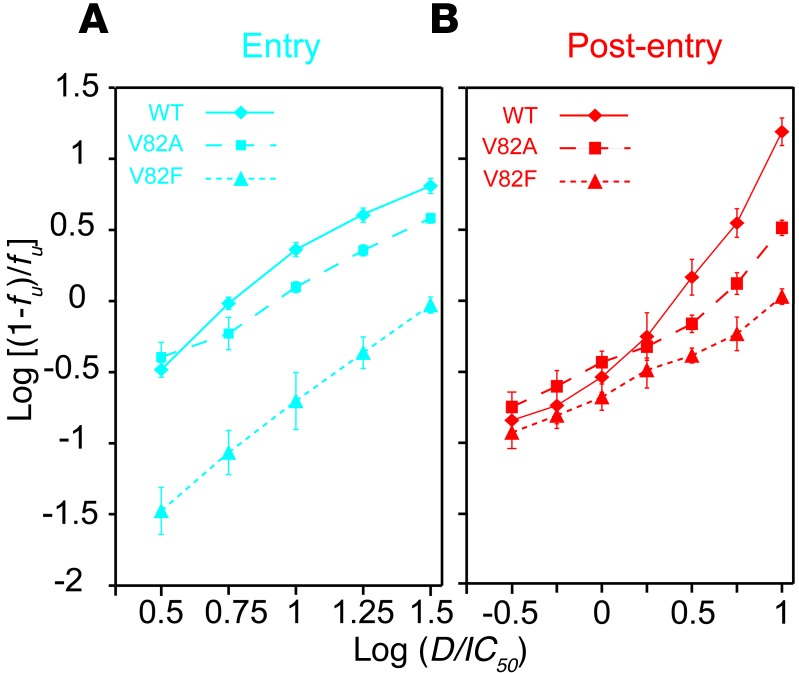
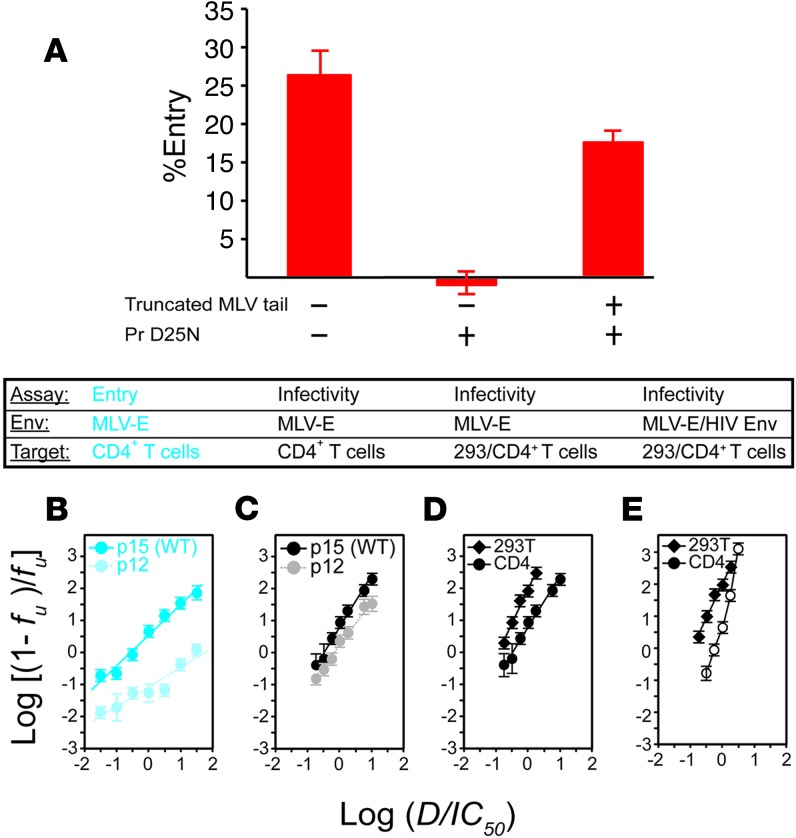
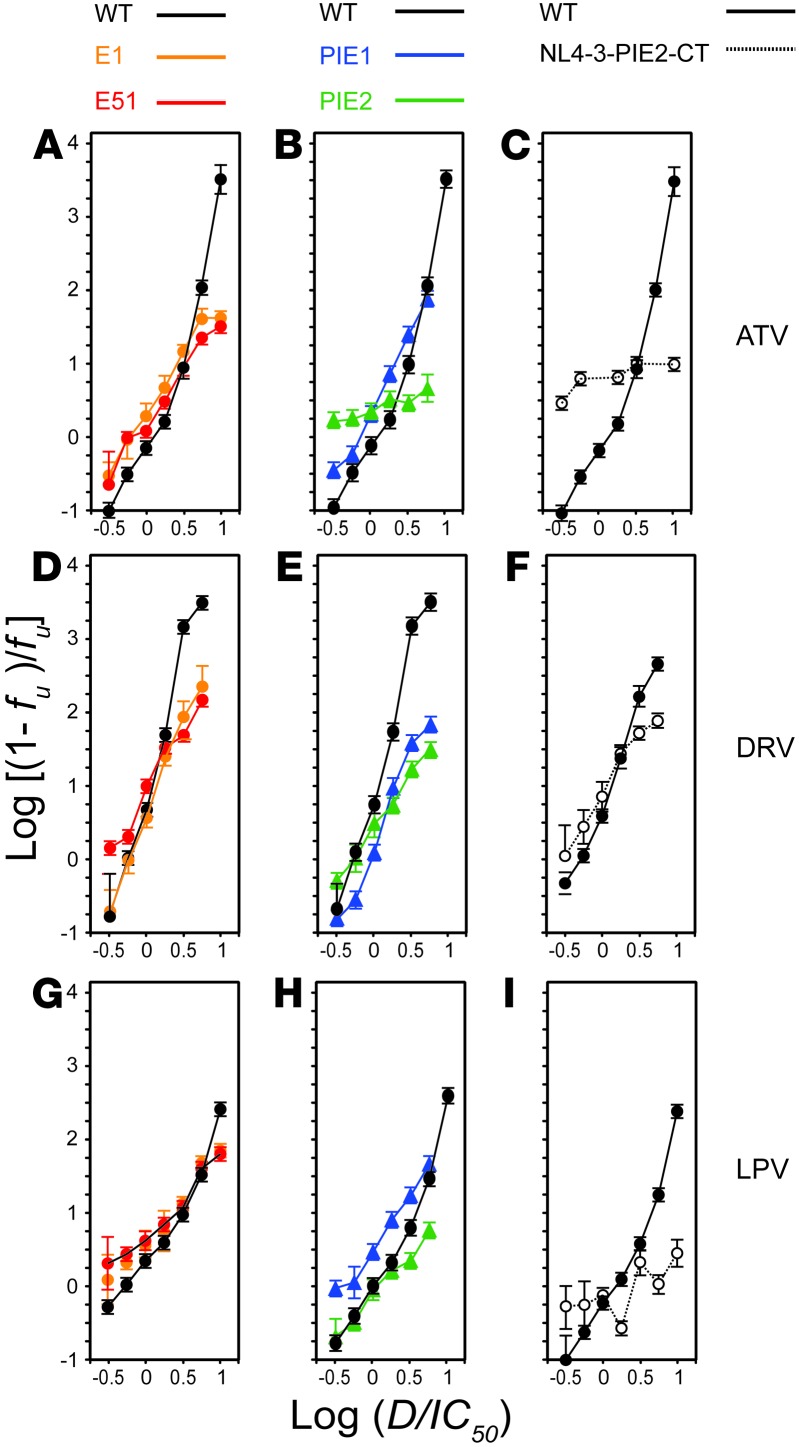
HIV-1 protease inhibitors (PIs) are among the most effective antiretroviral drugs. They are characterized by highly cooperative dose-response curves that are not explained by current pharmacodynamic theory. An unresolved problem affecting the clinical use of PIs is that patients who fail PI-containing regimens often have virus that lacks protease mutations, in apparent violation of fundamental evolutionary theory. Here, we show that these unresolved issues can be explained through analysis of the effects of PIs on distinct steps in the viral life cycle. We found that PIs do not affect virion release from infected cells but block entry, reverse transcription, and post-reverse transcription steps. The overall dose-response curves could be reconstructed by combining the curves for each step using the Bliss independence principle, showing that independent inhibition of multiple distinct steps in the life cycle generates the highly cooperative dose-response curves that make these drugs uniquely effective. Approximately half of the inhibitory potential of PIs is manifest at the entry step, likely reflecting interactions between the uncleaved Gag and the cytoplasmic tail (CT) of the Env protein. Sequence changes in the CT alone, which are ignored in current clinical tests for PI resistance, conferred PI resistance, providing an explanation for PI failure without resistance.
Figures
Comment in
-
Lack of protease inhibitor resistance following treatment failure--too good to be true?J Clin Invest. 2013 Sep;123(9):3704-5. doi: 10.1172/JCI71784. Epub 2013 Aug 27. J Clin Invest. 2013. PMID: 23979153 Free PMC article.
Similar articles
-
GRL-0519, a novel oxatricyclic ligand-containing nonpeptidic HIV-1 protease inhibitor (PI), potently suppresses replication of a wide spectrum of multi-PI-resistant HIV-1 variants in vitro.Antimicrob Agents Chemother. 2013 May;57(5):2036-46. doi: 10.1128/AAC.02189-12. Epub 2013 Feb 12. Antimicrob Agents Chemother. 2013. PMID: 23403426 Free PMC article.
-
Natural polymorphisms and unusual mutations in HIV-1 protease with potential antiretroviral resistance: a bioinformatic analysis.BMC Bioinformatics. 2014 Mar 15;15:72. doi: 10.1186/1471-2105-15-72. BMC Bioinformatics. 2014. PMID: 24629078 Free PMC article.
-
GRL-04810 and GRL-05010, difluoride-containing nonpeptidic HIV-1 protease inhibitors (PIs) that inhibit the replication of multi-PI-resistant HIV-1 in vitro and possess favorable lipophilicity that may allow blood-brain barrier penetration.Antimicrob Agents Chemother. 2013 Dec;57(12):6110-21. doi: 10.1128/AAC.01420-13. Epub 2013 Sep 30. Antimicrob Agents Chemother. 2013. PMID: 24080647 Free PMC article.
-
Darunavir: a review of its use in the management of HIV infection in adults.Drugs. 2009;69(4):477-503. doi: 10.2165/00003495-200969040-00007. Drugs. 2009. PMID: 19323590 Review.
-
The influence of protease inhibitor resistance profiles on selection of HIV therapy in treatment-naive patients.Antivir Ther. 2004 Jun;9(3):301-14. Antivir Ther. 2004. PMID: 15259893 Review.
Cited by
-
Nucleocapsid Protein: A Desirable Target for Future Therapies Against HIV-1.Curr Top Microbiol Immunol. 2015;389:53-92. doi: 10.1007/82_2015_433. Curr Top Microbiol Immunol. 2015. PMID: 25749978 Free PMC article. Review.
-
Effectiveness and safety of dual therapy with rilpivirine and boosted darunavir in treatment-experienced patients with advanced HIV infection: a preliminary 24 week analysis (RIDAR study).BMC Infect Dis. 2019 Feb 28;19(1):207. doi: 10.1186/s12879-019-3817-6. BMC Infect Dis. 2019. PMID: 30819101 Free PMC article.
-
Human Immunodeficiency Virus Type 1 Drug Resistance Mutations Update.J Infect Dis. 2017 Dec 1;216(suppl_9):S843-S846. doi: 10.1093/infdis/jix398. J Infect Dis. 2017. PMID: 28968669 Free PMC article.
-
Cross-clade simultaneous HIV drug resistance genotyping for reverse transcriptase, protease, and integrase inhibitor mutations by Illumina MiSeq.Retrovirology. 2014 Dec 23;11:122. doi: 10.1186/s12977-014-0122-8. Retrovirology. 2014. PMID: 25533166 Free PMC article.
-
HIV evolution and diversity in ART-treated patients.Retrovirology. 2018 Jan 30;15(1):14. doi: 10.1186/s12977-018-0395-4. Retrovirology. 2018. PMID: 29378595 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
