

Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene
- PMID: 23974870
- PMCID: PMC3874936
- DOI: 10.1038/ng.2745
Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene
Abstract
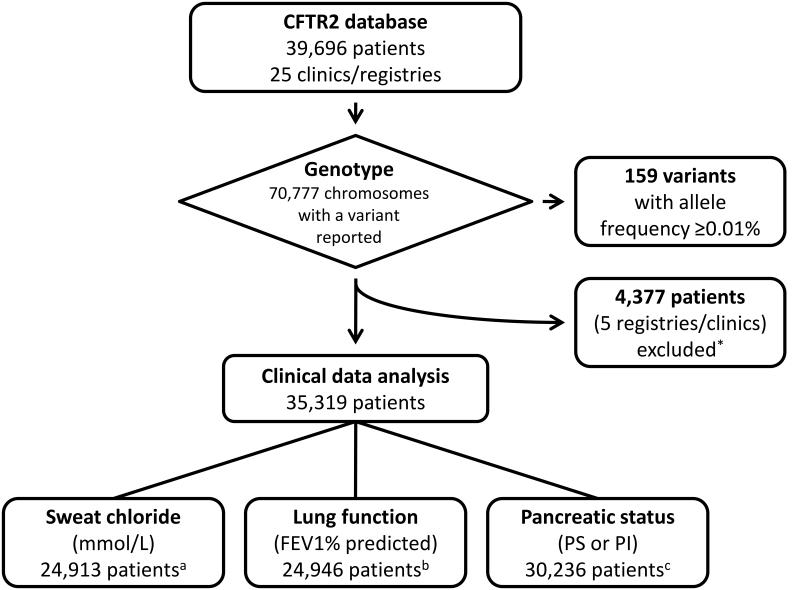
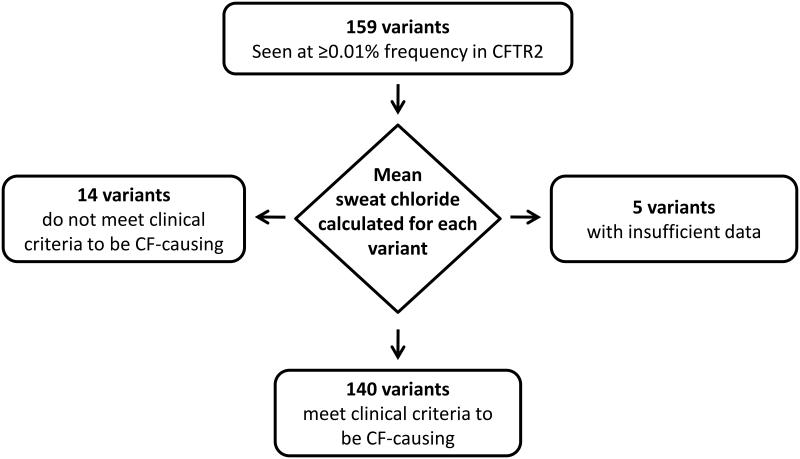
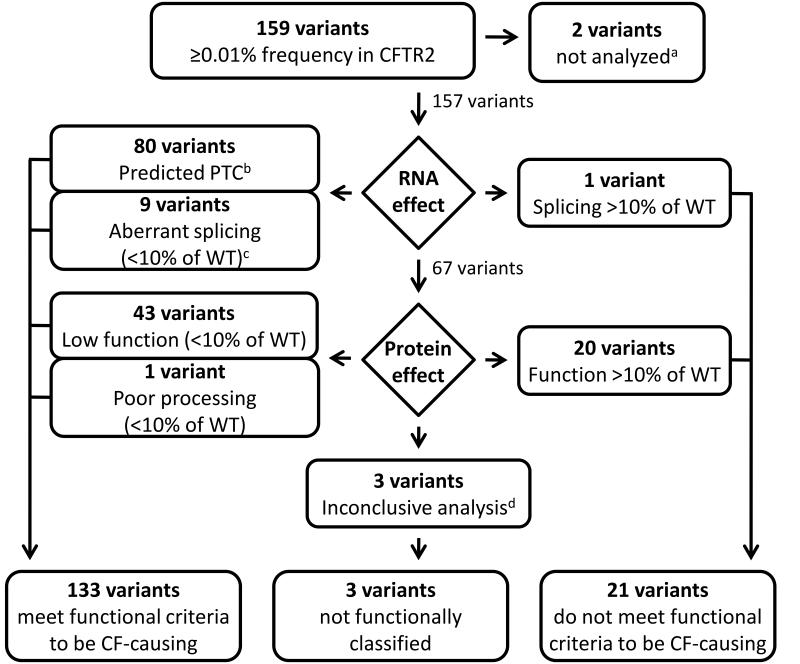
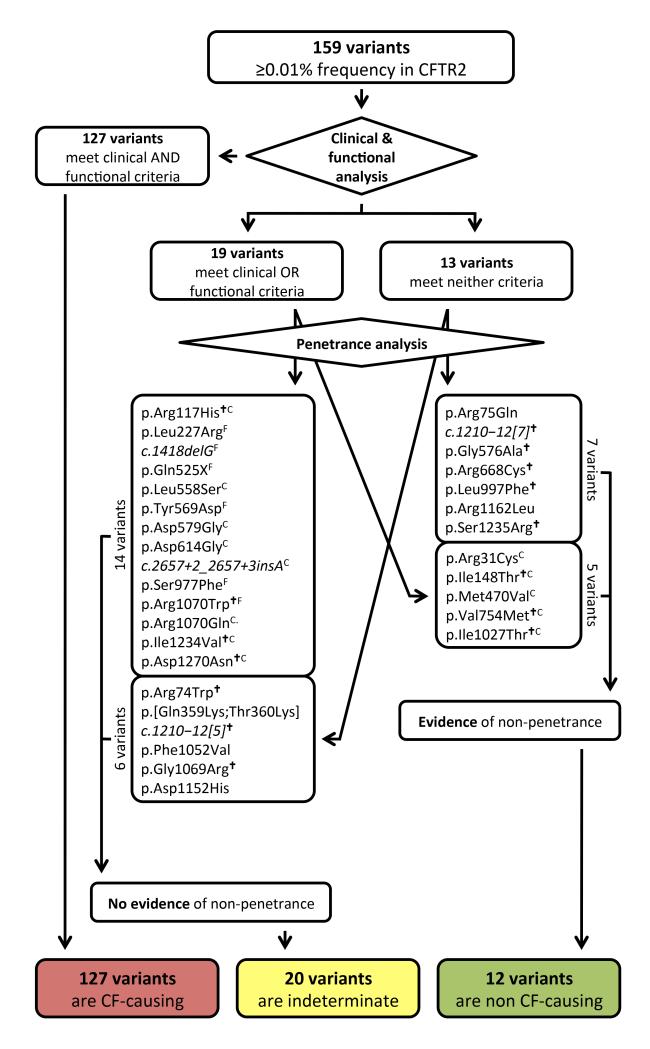
Allelic heterogeneity in disease-causing genes presents a substantial challenge to the translation of genomic variation into clinical practice. Few of the almost 2,000 variants in the cystic fibrosis transmembrane conductance regulator gene CFTR have empirical evidence that they cause cystic fibrosis. To address this gap, we collected both genotype and phenotype data for 39,696 individuals with cystic fibrosis in registries and clinics in North America and Europe. In these individuals, 159 CFTR variants had an allele frequency of ł0.01%. These variants were evaluated for both clinical severity and functional consequence, with 127 (80%) meeting both clinical and functional criteria consistent with disease. Assessment of disease penetrance in 2,188 fathers of individuals with cystic fibrosis enabled assignment of 12 of the remaining 32 variants as neutral, whereas the other 20 variants remained of indeterminate effect. This study illustrates that sourcing data directly from well-phenotyped subjects can address the gap in our ability to interpret clinically relevant genomic variation.
Figures
Similar articles
-
Applying Cystic Fibrosis Transmembrane Conductance Regulator Genetics and CFTR2 Data to Facilitate Diagnoses.J Pediatr. 2017 Feb;181S:S27-S32.e1. doi: 10.1016/j.jpeds.2016.09.063. J Pediatr. 2017. PMID: 28129809
-
Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis.Am J Respir Crit Care Med. 2019 May 1;199(9):1116-1126. doi: 10.1164/rccm.201901-0145OC. Am J Respir Crit Care Med. 2019. PMID: 30888834 Free PMC article.
-
Molecular Genetics of Cystic Fibrosis Transmembrane Conductance Regulator: Genotype and Phenotype.Pediatr Clin North Am. 2016 Aug;63(4):585-98. doi: 10.1016/j.pcl.2016.04.002. Pediatr Clin North Am. 2016. PMID: 27469177 Review.
-
Genetics of Cystic Fibrosis: Clinical Implications.Clin Chest Med. 2016 Mar;37(1):9-16. doi: 10.1016/j.ccm.2015.11.002. Epub 2015 Dec 24. Clin Chest Med. 2016. PMID: 26857764 Review.
-
R248G cystic fibrosis transmembrane conductance regulator mutation in three siblings presenting with recurrent acute pancreatitis and reproductive issues: a case series.J Med Case Rep. 2017 Feb 15;11(1):42. doi: 10.1186/s13256-016-1181-3. J Med Case Rep. 2017. PMID: 28196530 Free PMC article.
Cited by
-
Population Characteristics of the Spectrum and Frequencies of CFTR Gene Mutations in Patients with Cystic Fibrosis from the Republic of Bashkortostan (Russia).Genes (Basel). 2024 Oct 17;15(10):1335. doi: 10.3390/genes15101335. Genes (Basel). 2024. PMID: 39457459 Free PMC article.
-
Rescuing Trafficking Mutants of the ATP-binding Cassette Protein, ABCA4, with Small Molecule Correctors as a Treatment for Stargardt Eye Disease.J Biol Chem. 2015 Aug 7;290(32):19743-55. doi: 10.1074/jbc.M115.647685. Epub 2015 Jun 19. J Biol Chem. 2015. PMID: 26092729 Free PMC article.
-
Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells.Cells. 2020 Sep 13;9(9):2090. doi: 10.3390/cells9092090. Cells. 2020. PMID: 32933106 Free PMC article.
-
Analysis of cystic fibrosis-associated P67L CFTR illustrates barriers to personalized therapeutics for orphan diseases.JCI Insight. 2016 Sep 8;1(14):e86581. doi: 10.1172/jci.insight.86581. JCI Insight. 2016. PMID: 27660821 Free PMC article.
-
Initial Evaluation of Prospective and Parallel Assessments of Cystic Fibrosis Newborn Screening Protocols in Eastern Andalusia: IRT/IRT versus IRT/PAP/IRT.Int J Neonatal Screen. 2019 Sep 3;5(3):32. doi: 10.3390/ijns5030032. eCollection 2019 Sep. Int J Neonatal Screen. 2019. PMID: 33072991 Free PMC article.
References
-
- Chenevix-Trench G, et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–2027. - PubMed
-
- Richards CS, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008;10:294–300. - PubMed
-
- Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
