

Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis
- PMID: 23931993
- PMCID: PMC4411085
- DOI: 10.1016/j.neuron.2013.07.033
Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis
Abstract
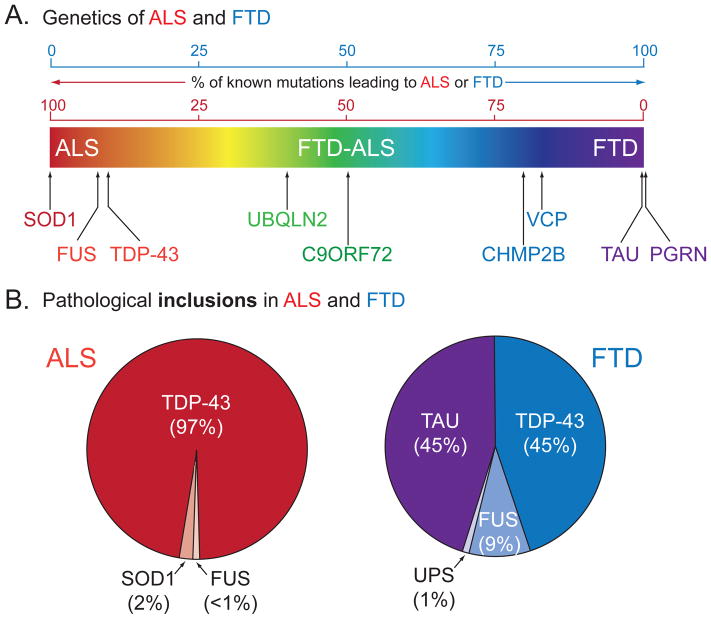
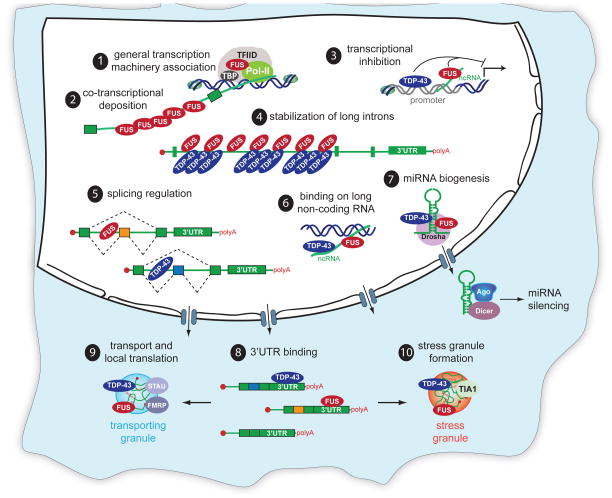
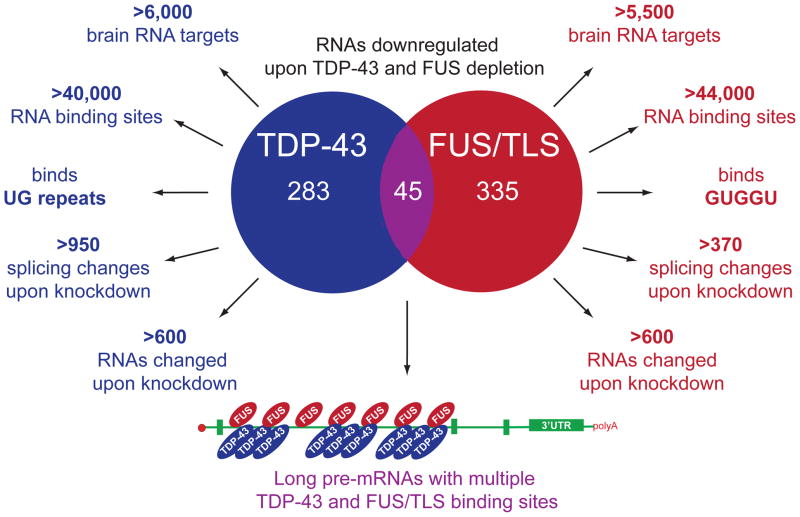
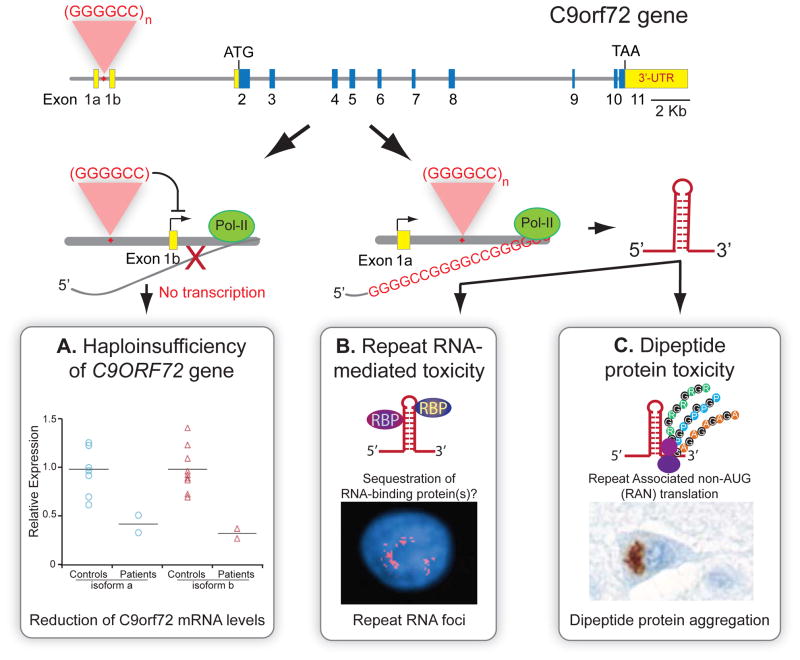
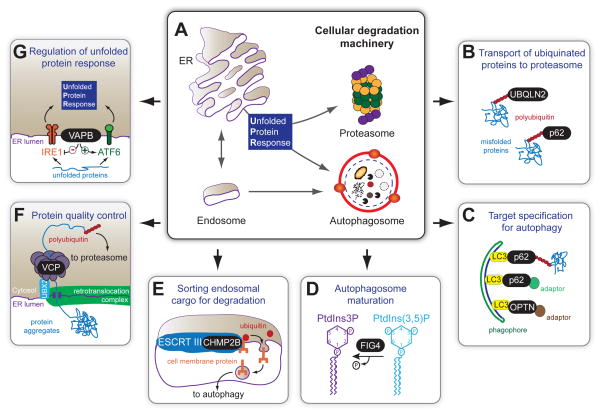
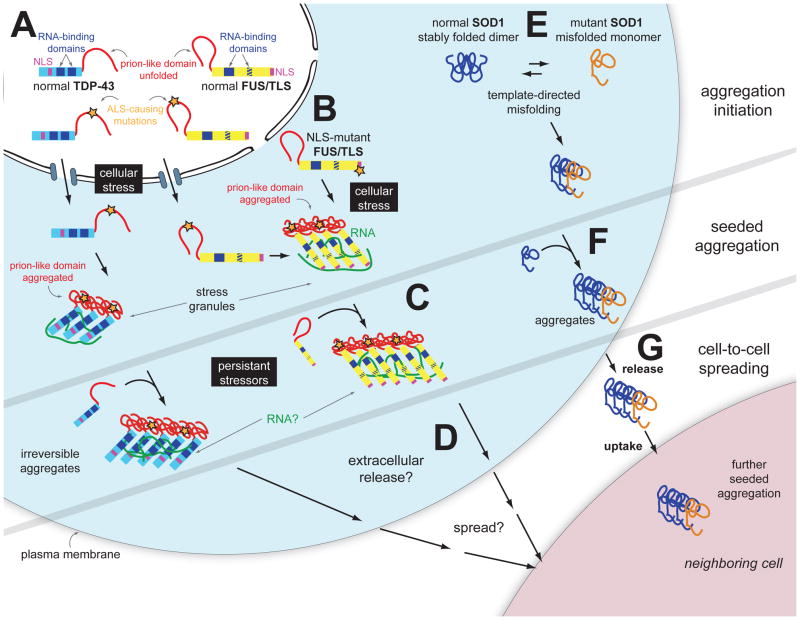
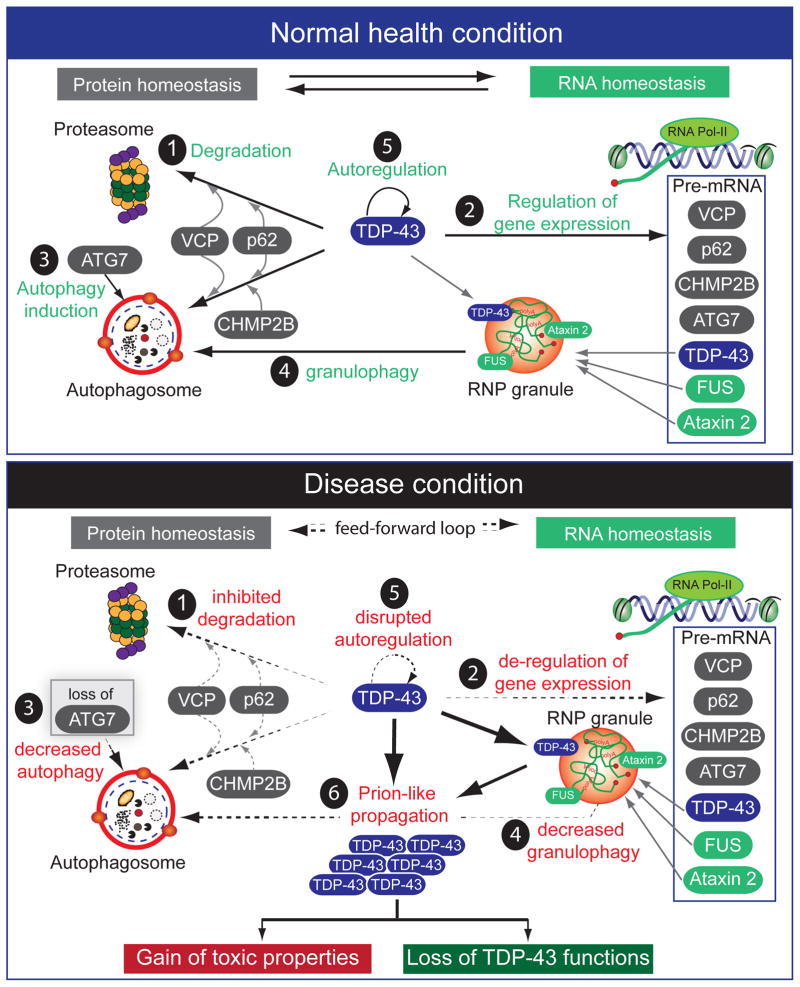
Breakthrough discoveries identifying common genetic causes for amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) have transformed our view of these disorders. They share unexpectedly similar signatures, including dysregulation in common molecular players including TDP-43, FUS/TLS, ubiquilin-2, VCP, and expanded hexanucleotide repeats within the C9ORF72 gene. Dysfunction in RNA processing and protein homeostasis is an emerging theme. We present the case here that these two processes are intimately linked, with disease-initiated perturbation of either leading to further deviation of both protein and RNA homeostasis through a feedforward loop including cell-to-cell prion-like spread that may represent the mechanism for relentless disease progression.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs.Nat Neurosci. 2012 Nov;15(11):1488-97. doi: 10.1038/nn.3230. Epub 2012 Sep 30. Nat Neurosci. 2012. PMID: 23023293 Free PMC article.
-
Making connections: pathology and genetics link amyotrophic lateral sclerosis with frontotemporal lobe dementia.J Mol Neurosci. 2011 Nov;45(3):663-75. doi: 10.1007/s12031-011-9637-9. Epub 2011 Sep 7. J Mol Neurosci. 2011. PMID: 21901496 Review.
-
Amyotrophic lateral sclerosis: an update on recent genetic insights.J Neurol. 2013 Nov;260(11):2917-27. doi: 10.1007/s00415-013-7112-y. Epub 2013 Oct 2. J Neurol. 2013. PMID: 24085347 Review.
-
Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion.Acta Neuropathol. 2012 Jun;123(6):825-39. doi: 10.1007/s00401-012-0970-z. Epub 2012 Mar 18. Acta Neuropathol. 2012. PMID: 22426854 Free PMC article.
-
Hippocampal aggregation signatures of pathogenic UBQLN2 in amyotrophic lateral sclerosis and frontotemporal dementia.Brain. 2024 Oct 3;147(10):3547-3561. doi: 10.1093/brain/awae140. Brain. 2024. PMID: 38703371 Free PMC article.
Cited by
-
The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains.Elife. 2016 Sep 13;5:e17820. doi: 10.7554/eLife.17820. Elife. 2016. PMID: 27623008 Free PMC article.
-
NOS1AP is a novel molecular target and critical factor in TDP-43 pathology.Brain Commun. 2022 Sep 23;4(5):fcac242. doi: 10.1093/braincomms/fcac242. eCollection 2022. Brain Commun. 2022. PMID: 36267332 Free PMC article.
-
Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43.Acta Neuropathol. 2015 Nov;130(5):643-60. doi: 10.1007/s00401-015-1460-x. Epub 2015 Jul 22. Acta Neuropathol. 2015. PMID: 26197969 Free PMC article.
-
Variability in Clinical Phenotype in TARDBP Mutations: Amyotrophic Lateral Sclerosis Case Description and Literature Review.Genes (Basel). 2023 Nov 4;14(11):2039. doi: 10.3390/genes14112039. Genes (Basel). 2023. PMID: 38002982 Free PMC article.
-
FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP.Proc Natl Acad Sci U S A. 2015 Jul 14;112(28):8608-13. doi: 10.1073/pnas.1506282112. Epub 2015 Jun 29. Proc Natl Acad Sci U S A. 2015. PMID: 26124092 Free PMC article.
References
-
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobágyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. - PubMed
-
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–436. - PubMed
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. - PubMed
-
- Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA. 2013;110:E736–745. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
