

Directional RNA-seq reveals highly complex condition-dependent transcriptomes in E. coli K12 through accurate full-length transcripts assembling
- PMID: 23899370
- PMCID: PMC3734233
- DOI: 10.1186/1471-2164-14-520
Directional RNA-seq reveals highly complex condition-dependent transcriptomes in E. coli K12 through accurate full-length transcripts assembling
Abstract
Background: Although prokaryotic gene transcription has been studied over decades, many aspects of the process remain poorly understood. Particularly, recent studies have revealed that transcriptomes in many prokaryotes are far more complex than previously thought. Genes in an operon are often alternatively and dynamically transcribed under different conditions, and a large portion of genes and intergenic regions have antisense RNA (asRNA) and non-coding RNA (ncRNA) transcripts, respectively. Ironically, similar studies have not been conducted in the model bacterium E coli K12, thus it is unknown whether or not the bacterium possesses similar complex transcriptomes. Furthermore, although RNA-seq becomes the major method for analyzing the complexity of prokaryotic transcriptome, it is still a challenging task to accurately assemble full length transcripts using short RNA-seq reads.
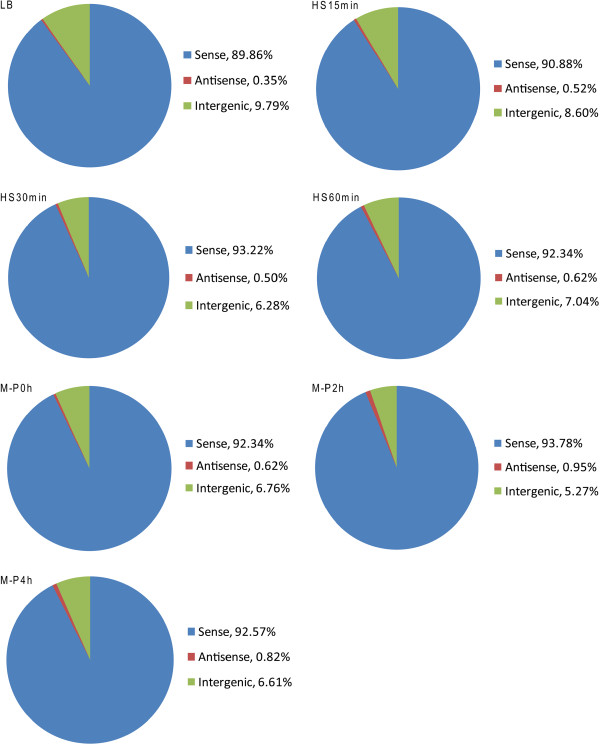
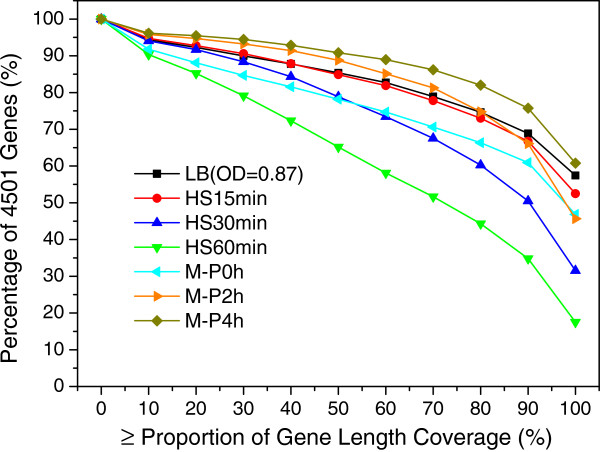
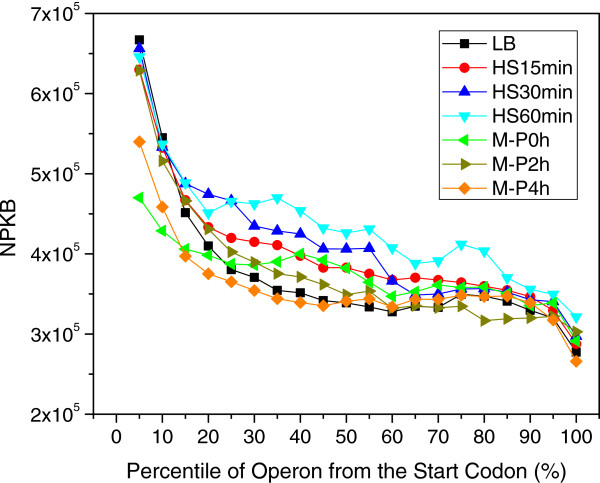
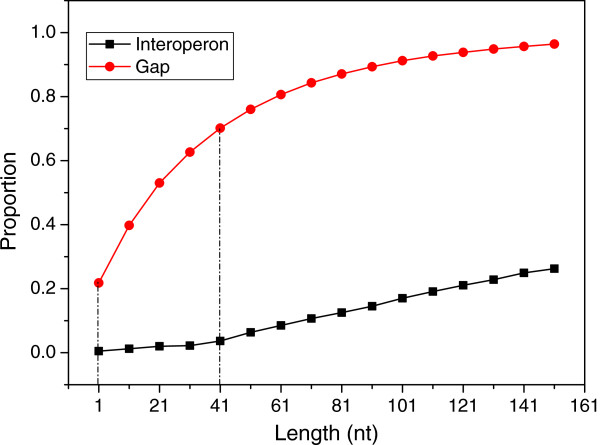
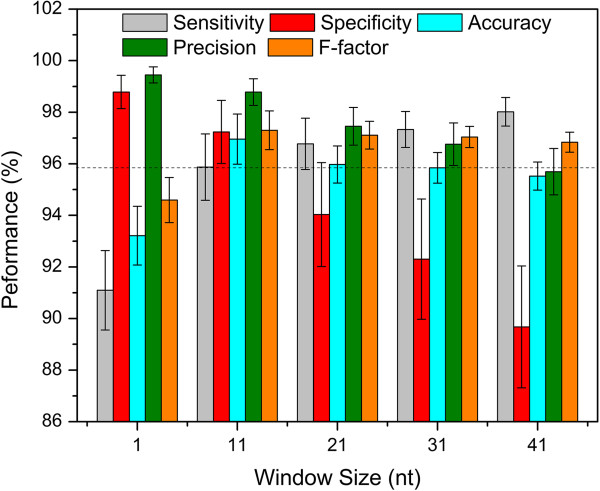
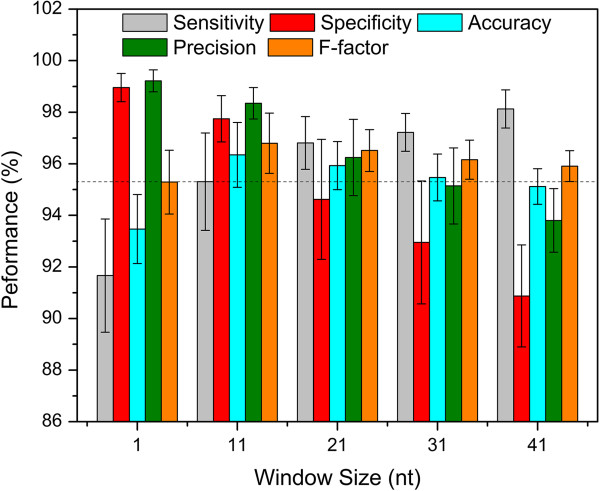
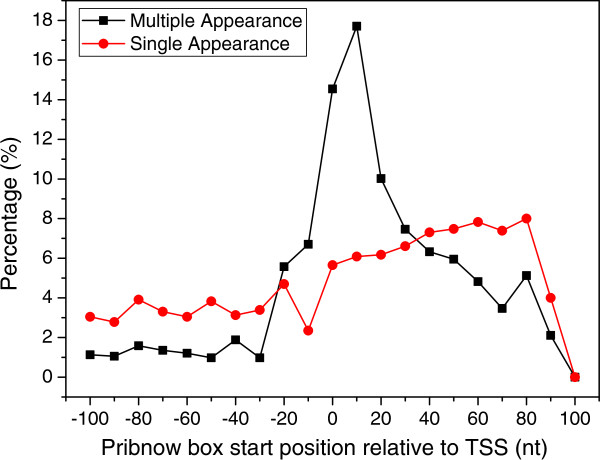
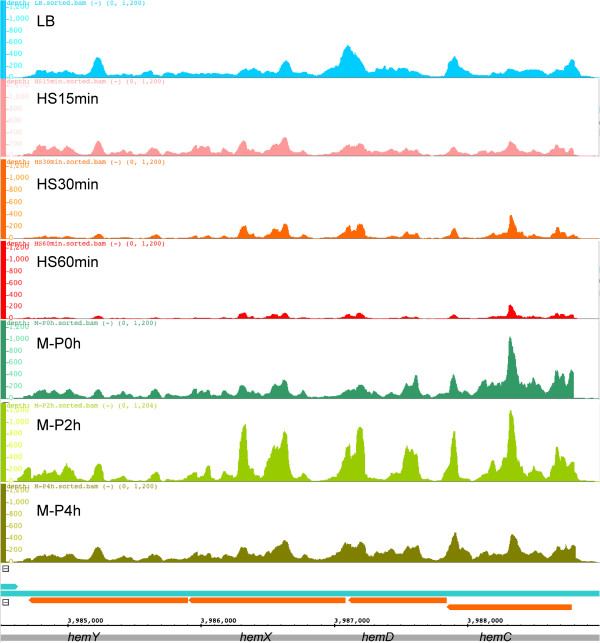
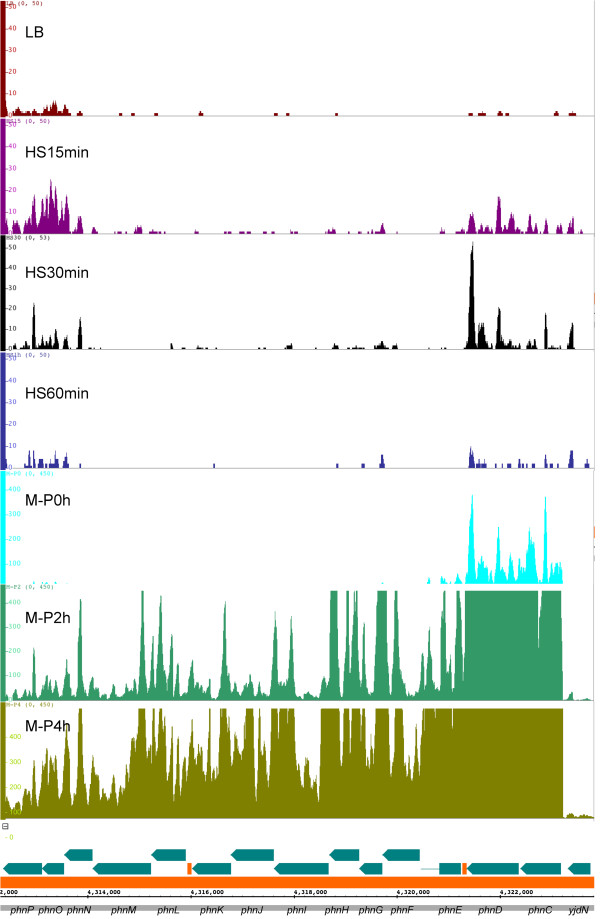
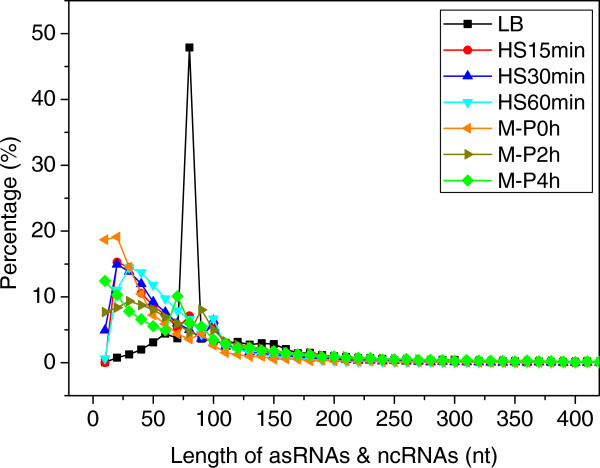
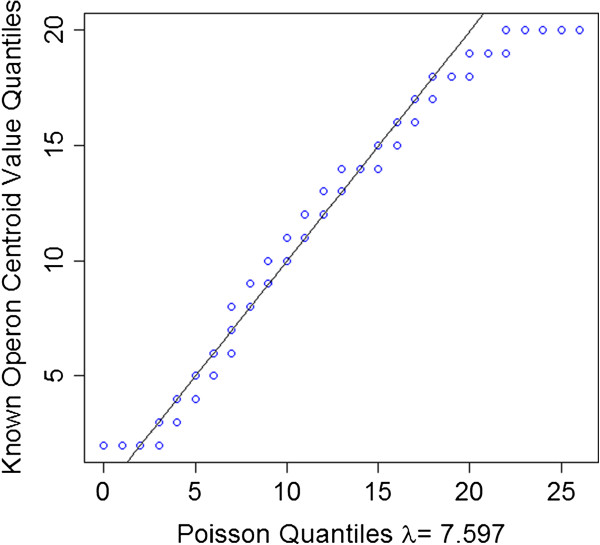
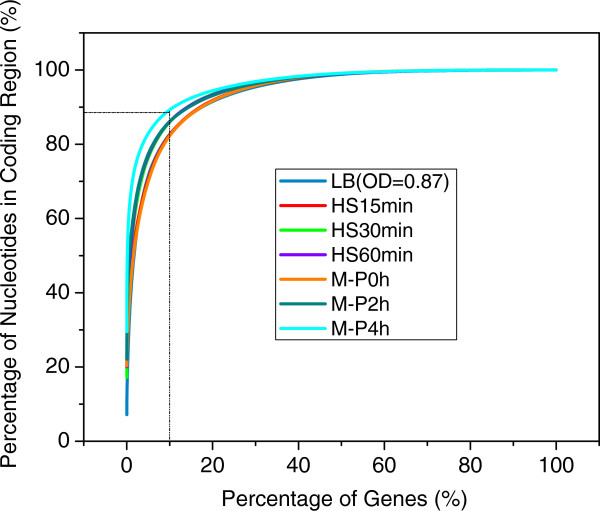
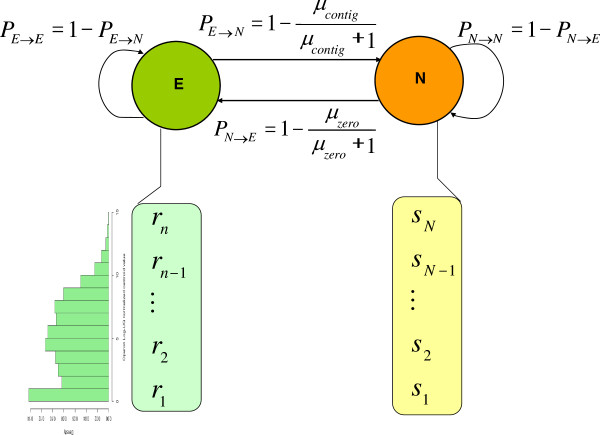
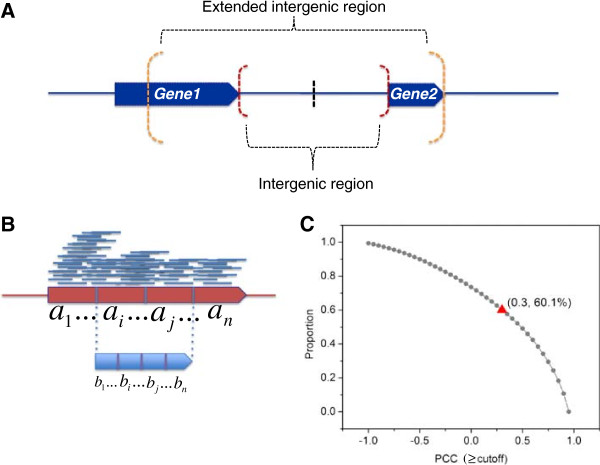
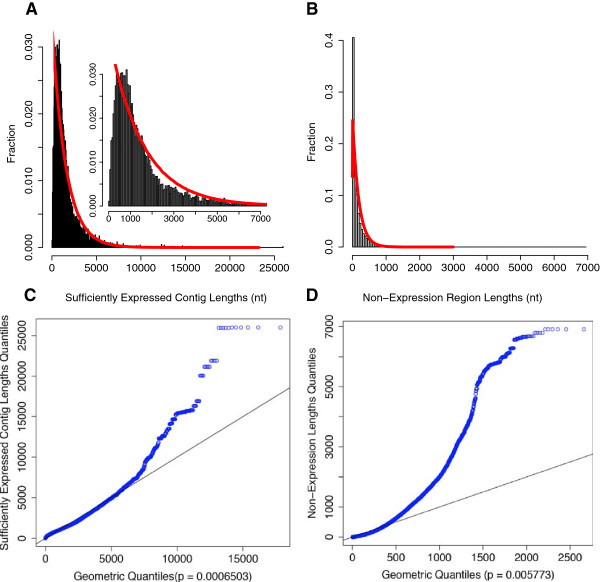
Results: To fill these gaps, we have profiled the transcriptomes of E. coli K12 under different culture conditions and growth phases using a highly specific directional RNA-seq technique that can capture various types of transcripts in the bacterial cells, combined with a highly accurate and robust algorithm and tool TruHMM (http://bioinfolab.uncc.edu/TruHmm_package/) for assembling full length transcripts. We found that 46.9 ~ 63.4% of expressed operons were utilized in their putative alternative forms, 72.23 ~ 89.54% genes had putative asRNA transcripts and 51.37 ~ 72.74% intergenic regions had putative ncRNA transcripts under different culture conditions and growth phases.
Conclusions: As has been demonstrated in many other prokaryotes, E. coli K12 also has a highly complex and dynamic transcriptomes under different culture conditions and growth phases. Such complex and dynamic transcriptomes might play important roles in the physiology of the bacterium. TruHMM is a highly accurate and robust algorithm for assembling full-length transcripts in prokaryotes using directional RNA-seq short reads.
Figures
Similar articles
-
Antisense transcription and its roles in adaption to environmental stress in E. coli.bioRxiv [Preprint]. 2023 Mar 24:2023.03.23.533988. doi: 10.1101/2023.03.23.533988. bioRxiv. 2023. PMID: 36993172 Free PMC article. Preprint.
-
Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing.mBio. 2014 Jul 8;5(4):e01442-14. doi: 10.1128/mBio.01442-14. mBio. 2014. PMID: 25006232 Free PMC article.
-
Deciphering transcript architectural complexity in bacteria and archaea.mBio. 2024 Oct 16;15(10):e0235924. doi: 10.1128/mbio.02359-24. Epub 2024 Sep 17. mBio. 2024. PMID: 39287442 Free PMC article.
-
Decoding co-/post-transcriptional complexities of plant transcriptomes and epitranscriptome using next-generation sequencing technologies.Biochem Soc Trans. 2020 Dec 18;48(6):2399-2414. doi: 10.1042/BST20190492. Biochem Soc Trans. 2020. PMID: 33196096 Review.
-
The complexity of bacterial transcriptomes.J Biotechnol. 2016 Aug 20;232:69-78. doi: 10.1016/j.jbiotec.2015.09.041. Epub 2015 Oct 9. J Biotechnol. 2016. PMID: 26450562 Review.
Cited by
-
Differentially Expressed Genes of Pseudomonas aeruginosa Isolates from Eyes with Keratitis and Healthy Conjunctival Sacs.Infect Drug Resist. 2022 Aug 12;15:4495-4506. doi: 10.2147/IDR.S374335. eCollection 2022. Infect Drug Resist. 2022. PMID: 35983295 Free PMC article.
-
Gene finding in metatranscriptomic sequences.BMC Bioinformatics. 2014;15 Suppl 9(Suppl 9):S8. doi: 10.1186/1471-2105-15-S9-S8. Epub 2014 Sep 10. BMC Bioinformatics. 2014. PMID: 25253067 Free PMC article.
-
An assessment of bacterial small RNA target prediction programs.RNA Biol. 2015;12(5):509-13. doi: 10.1080/15476286.2015.1020269. RNA Biol. 2015. PMID: 25760244 Free PMC article. Review.
-
Distinguishing between productive and abortive promoters using a random forest classifier in Mycoplasma pneumoniae.Nucleic Acids Res. 2015 Apr 20;43(7):3442-53. doi: 10.1093/nar/gkv170. Epub 2015 Mar 16. Nucleic Acids Res. 2015. PMID: 25779052 Free PMC article.
-
Revisiting operons: an analysis of the landscape of transcriptional units in E. coli.BMC Bioinformatics. 2015 Nov 4;16:356. doi: 10.1186/s12859-015-0805-8. BMC Bioinformatics. 2015. PMID: 26538447 Free PMC article.
References
-
- Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T. et al.EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011;39:D583–590. doi: 10.1093/nar/gkq1143. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
