

Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species
- PMID: 23870653
- PMCID: PMC3844414
- DOI: 10.1186/2047-217X-2-10
Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species
Abstract
Background: The process of generating raw genome sequence data continues to become cheaper, faster, and more accurate. However, assembly of such data into high-quality, finished genome sequences remains challenging. Many genome assembly tools are available, but they differ greatly in terms of their performance (speed, scalability, hardware requirements, acceptance of newer read technologies) and in their final output (composition of assembled sequence). More importantly, it remains largely unclear how to best assess the quality of assembled genome sequences. The Assemblathon competitions are intended to assess current state-of-the-art methods in genome assembly.
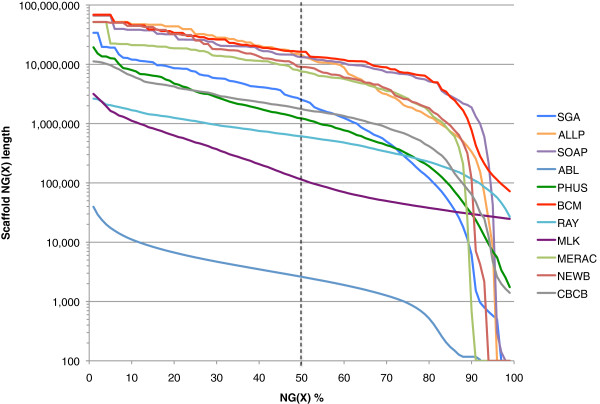
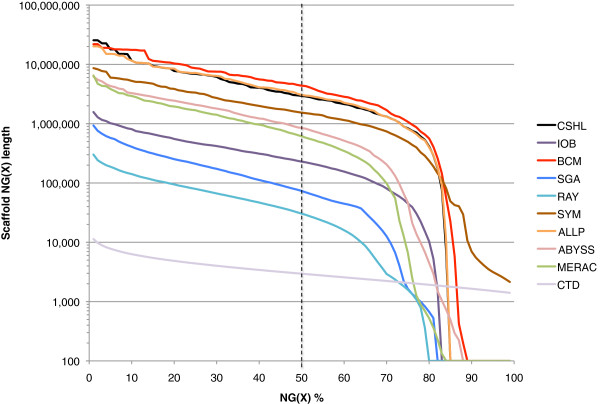
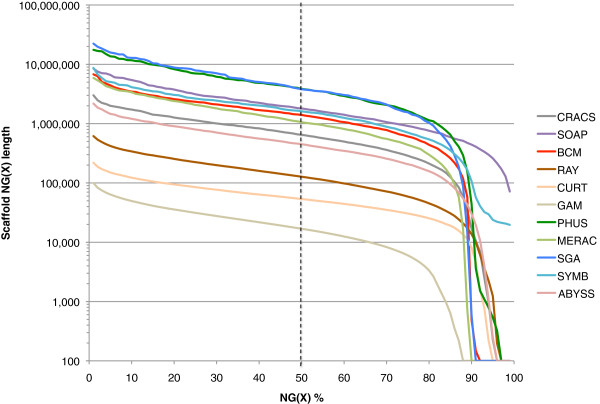
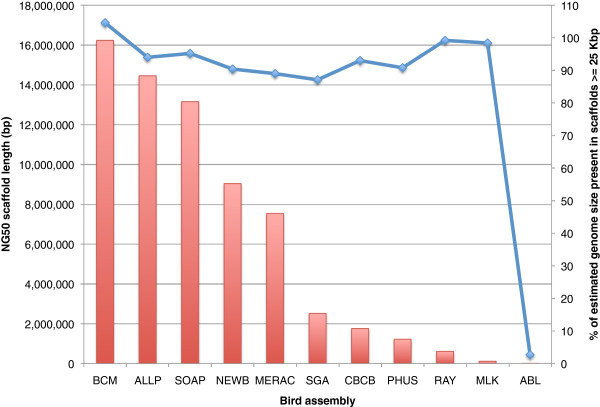
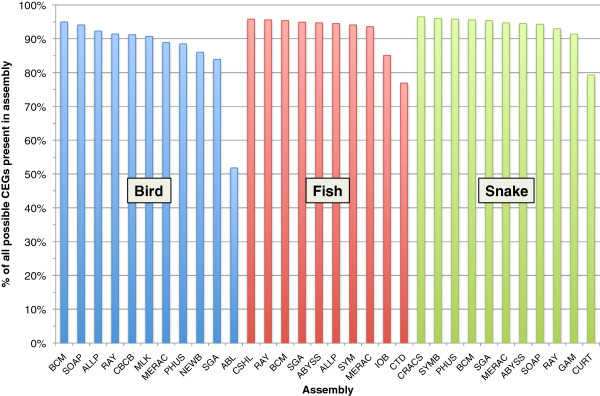
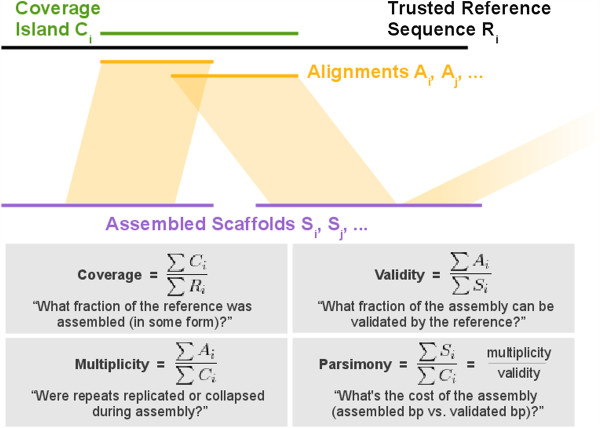
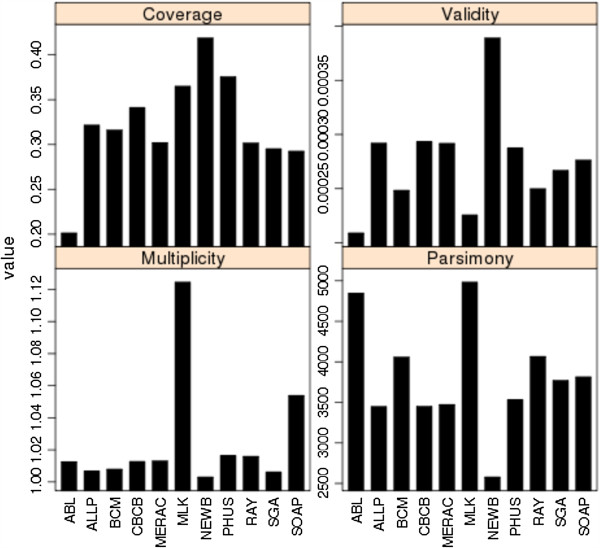
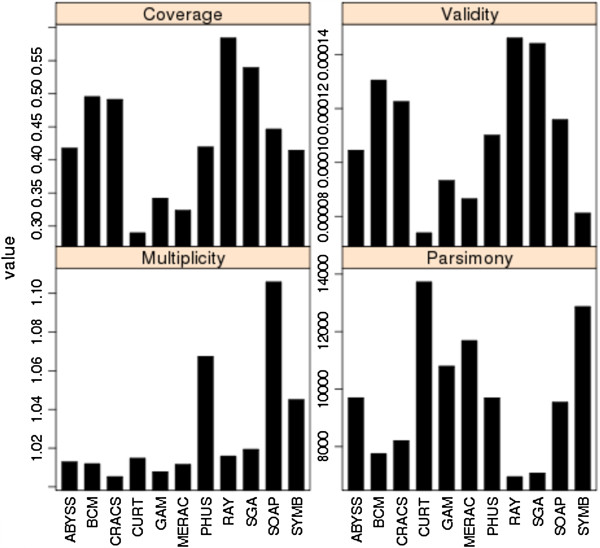
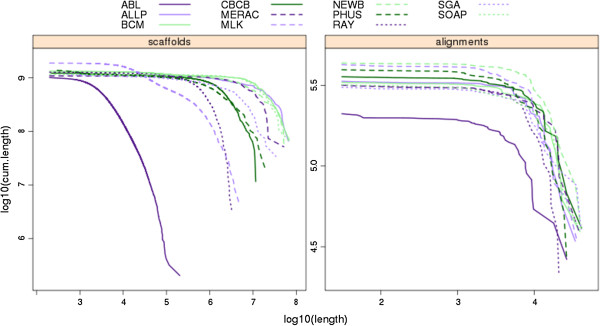
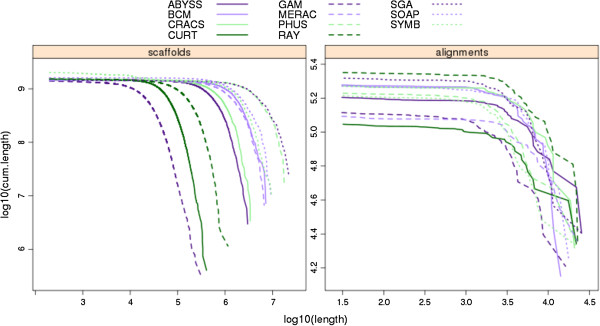
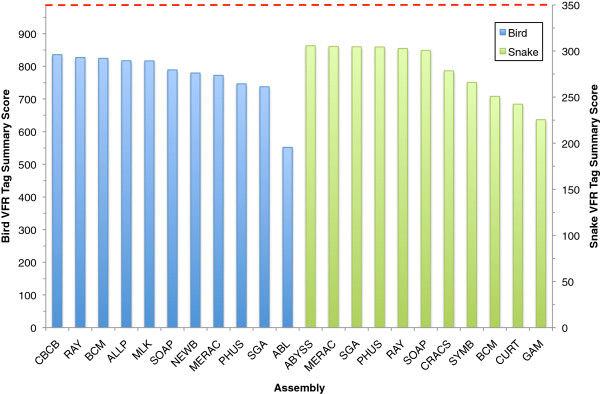
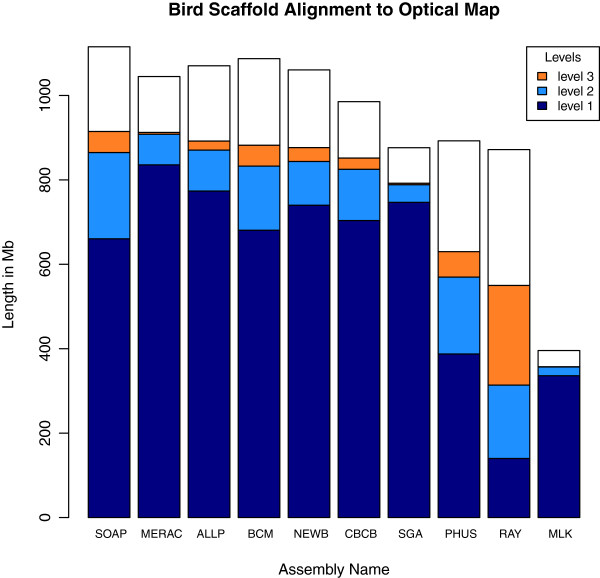
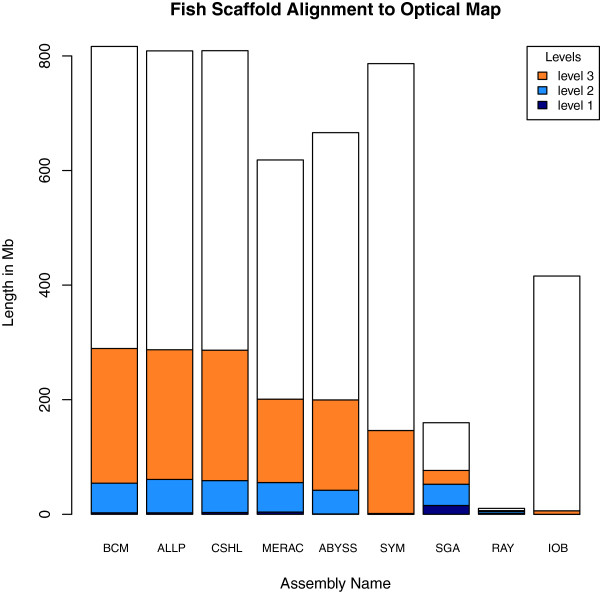
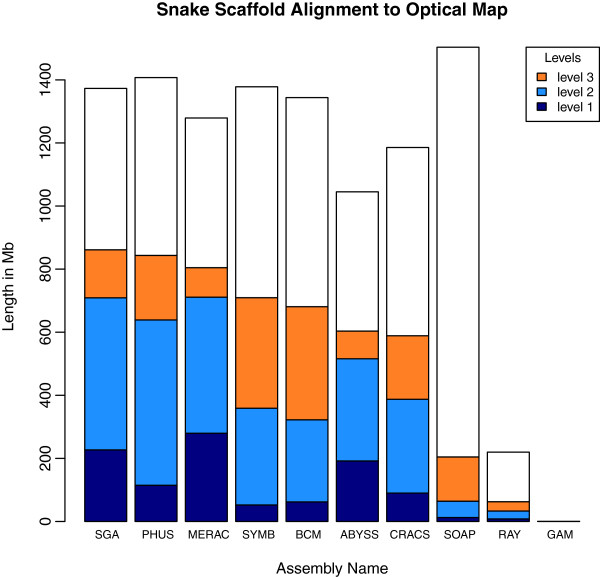
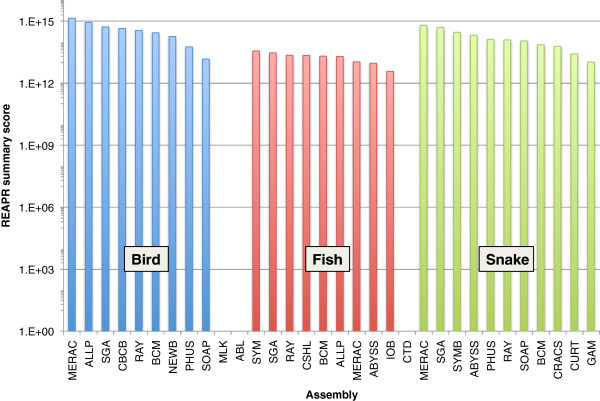
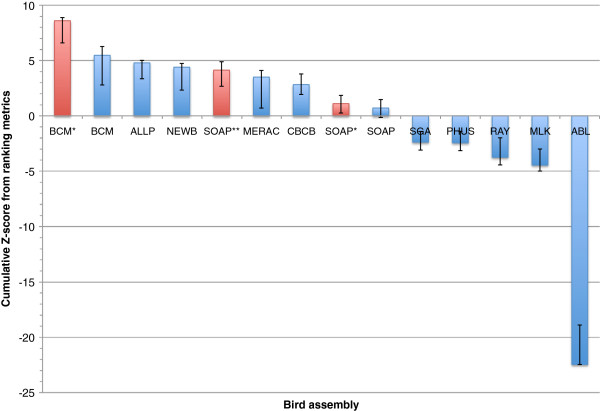
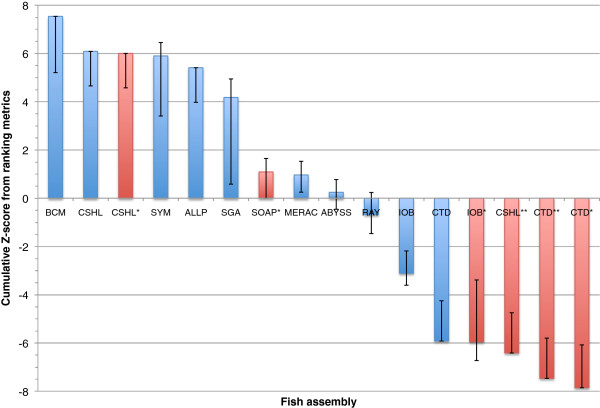
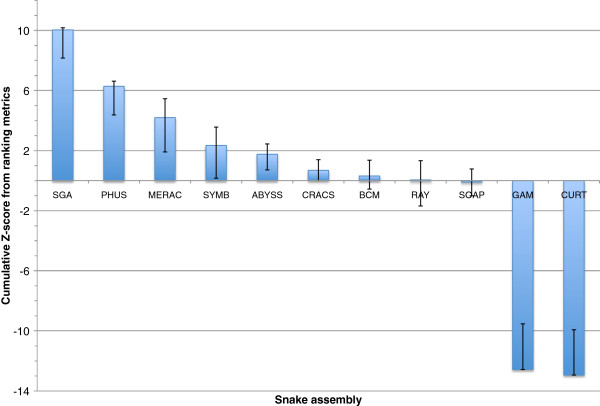
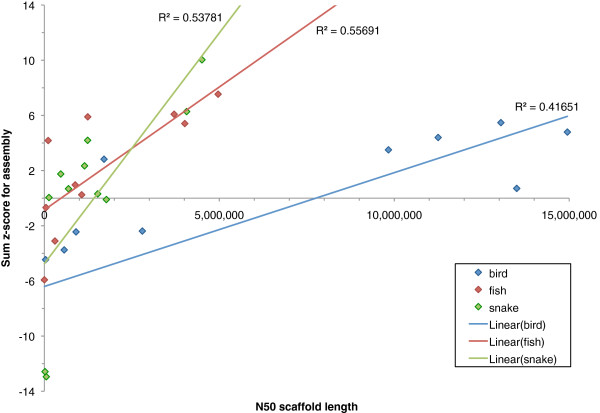
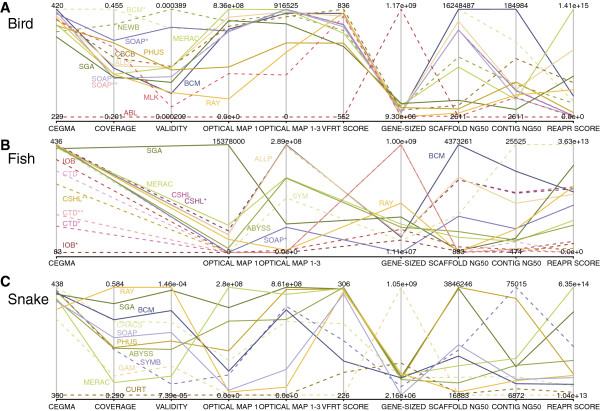
Results: In Assemblathon 2, we provided a variety of sequence data to be assembled for three vertebrate species (a bird, a fish, and snake). This resulted in a total of 43 submitted assemblies from 21 participating teams. We evaluated these assemblies using a combination of optical map data, Fosmid sequences, and several statistical methods. From over 100 different metrics, we chose ten key measures by which to assess the overall quality of the assemblies.
Conclusions: Many current genome assemblers produced useful assemblies, containing a significant representation of their genes and overall genome structure. However, the high degree of variability between the entries suggests that there is still much room for improvement in the field of genome assembly and that approaches which work well in assembling the genome of one species may not necessarily work well for another.
Figures

Similar articles
-
De novo likelihood-based measures for comparing genome assemblies.BMC Res Notes. 2013 Aug 22;6:334. doi: 10.1186/1756-0500-6-334. BMC Res Notes. 2013. PMID: 23965294 Free PMC article.
-
Assemblathon 1: a competitive assessment of de novo short read assembly methods.Genome Res. 2011 Dec;21(12):2224-41. doi: 10.1101/gr.126599.111. Epub 2011 Sep 16. Genome Res. 2011. PMID: 21926179 Free PMC article.
-
Fragmentation and Coverage Variation in Viral Metagenome Assemblies, and Their Effect in Diversity Calculations.Front Bioeng Biotechnol. 2015 Sep 17;3:141. doi: 10.3389/fbioe.2015.00141. eCollection 2015. Front Bioeng Biotechnol. 2015. PMID: 26442255 Free PMC article.
-
Evaluation of genome scaffolding tools using pooled clone sequencing.Turk J Biol. 2018 Dec 10;42(6):471-476. doi: 10.3906/biy-1805-42. eCollection 2018. Turk J Biol. 2018. PMID: 30983868 Free PMC article.
-
Toward a statistically explicit understanding of de novo sequence assembly.Bioinformatics. 2013 Dec 1;29(23):2959-63. doi: 10.1093/bioinformatics/btt525. Epub 2013 Sep 10. Bioinformatics. 2013. PMID: 24021385 Review.
Cited by
-
A whole-genome shotgun approach for assembling and anchoring the hexaploid bread wheat genome.Genome Biol. 2015 Jan 31;16(1):26. doi: 10.1186/s13059-015-0582-8. Genome Biol. 2015. PMID: 25637298 Free PMC article.
-
Genome Reduction and Secondary Metabolism of the Marine Sponge-Associated Cyanobacterium Leptothoe.Mar Drugs. 2021 May 24;19(6):298. doi: 10.3390/md19060298. Mar Drugs. 2021. PMID: 34073758 Free PMC article.
-
Genome-Wide Survey and Development of the First Microsatellite Markers Database (AnCorDB) in Anemone coronaria L.Int J Mol Sci. 2022 Mar 14;23(6):3126. doi: 10.3390/ijms23063126. Int J Mol Sci. 2022. PMID: 35328546 Free PMC article.
-
Novel Virus Identification through Metagenomics: A Systematic Review.Life (Basel). 2022 Dec 7;12(12):2048. doi: 10.3390/life12122048. Life (Basel). 2022. PMID: 36556413 Free PMC article. Review.
-
Ebola virus, but not Marburg virus, replicates efficiently and without required adaptation in snake cells.Virus Evol. 2018 Nov 28;4(2):vey034. doi: 10.1093/ve/vey034. eCollection 2018 Jul. Virus Evol. 2018. PMID: 30524754 Free PMC article.
References
-
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. - PubMed
-
- i5K - ArthropodBase wiki. http://www.arthropodgenomes.org/wiki/i5K.
