

Re-Directing CD4(+) T Cell Responses with the Flanking Residues of MHC Class II-Bound Peptides: The Core is Not Enough
- PMID: 23847615
- PMCID: PMC3696884
- DOI: 10.3389/fimmu.2013.00172
Re-Directing CD4(+) T Cell Responses with the Flanking Residues of MHC Class II-Bound Peptides: The Core is Not Enough
Abstract
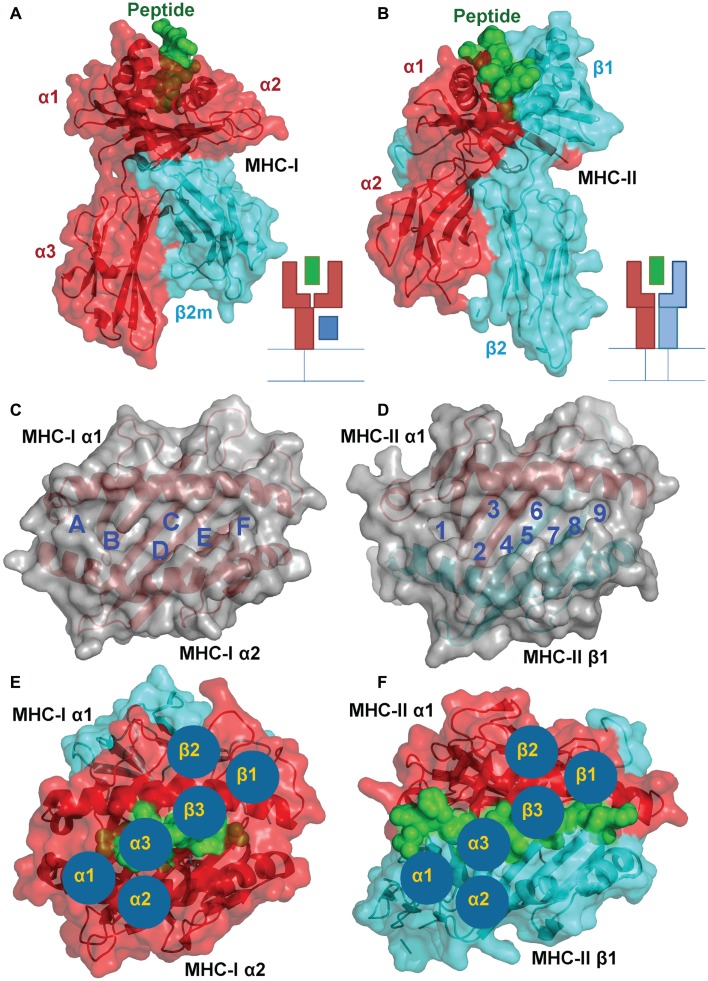
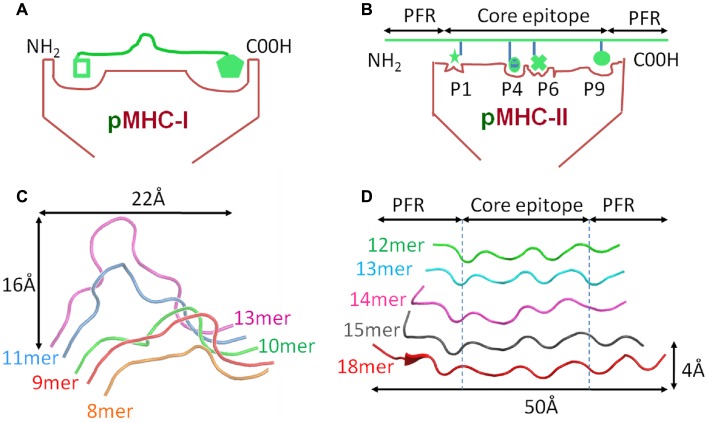
Recombinant αβ T cell receptors, expressed on T cell membranes, recognize short peptides presented at the cell surface in complex with MHC molecules. There are two main subsets of αβ T cells: CD8(+) T cells that recognize mainly cytosol-derived peptides in the context of MHC class I (pMHC-I), and CD4(+) T cells that recognize peptides usually derived from exogenous proteins presented by MHC class II (pMHC-II). Unlike the more uniform peptide lengths (usually 8-13mers) bound in the MHC-I closed groove, MHC-II presented peptides are of a highly variable length. The bound peptides consist of a core bound 9mer (reflecting the binding motif for the particular MHC-II type) but with variable peptide flanking residues (PFRs) that can extend from both the N- and C-terminus of the MHC-II binding groove. Although pMHC-I and pMHC-II play a virtually identical role during T cell responses (T cell antigen presentation) and are very similar in overall conformation, there exist a number of subtle but important differences that may govern the functional dichotomy observed between CD8(+) and CD4(+) T cells. Here, we provide an overview of the impact of structural differences between pMHC-I and pMHC-II and the molecular interactions with the T cell receptor including the functional importance of MHC-II PFRs. We consider how factors such as anatomical location, inflammatory milieu, and particular types of antigen presenting cell might, in theory, contribute to the quantitative (i.e., pMHC ligand frequency) as well as qualitative (i.e., variable PFR) nature of peptide epitopes, and hence offer a means of control and influence of a CD4(+) T cell response. Lastly, we review our recent findings showing how modifications to MHC-II PFRs can modify CD4(+) T cell antigen recognition. These findings may have novel applications for the development of CD4(+) T cell peptide vaccines and diagnostics.
Keywords: MHC processing; T cell receptor; T cell repertoire; crystal structure; modified peptide; peptide flanking residue; peptide-major histocompatibility complex class II; vaccine.
Figures



Similar articles
-
Enhanced Detection of Antigen-Specific CD4+ T Cells Using Altered Peptide Flanking Residue Peptide-MHC Class II Multimers.J Immunol. 2015 Dec 15;195(12):5827-36. doi: 10.4049/jimmunol.1402787. Epub 2015 Nov 9. J Immunol. 2015. PMID: 26553072 Free PMC article.
-
Structural and biophysical insights into the role of CD4 and CD8 in T cell activation.Front Immunol. 2013 Jul 22;4:206. doi: 10.3389/fimmu.2013.00206. eCollection 2013. Front Immunol. 2013. PMID: 23885256 Free PMC article.
-
Naturally processed HLA class II peptides reveal highly conserved immunogenic flanking region sequence preferences that reflect antigen processing rather than peptide-MHC interactions.J Immunol. 2001 Jun 1;166(11):6720-7. doi: 10.4049/jimmunol.166.11.6720. J Immunol. 2001. PMID: 11359828
-
The Role of Molecular Flexibility in Antigen Presentation and T Cell Receptor-Mediated Signaling.Front Immunol. 2018 Jul 17;9:1657. doi: 10.3389/fimmu.2018.01657. eCollection 2018. Front Immunol. 2018. PMID: 30065727 Free PMC article. Review.
-
MHC Molecules, T cell Receptors, Natural Killer Cell Receptors, and Viral Immunoevasins-Key Elements of Adaptive and Innate Immunity.Adv Exp Med Biol. 2019;1172:21-62. doi: 10.1007/978-981-13-9367-9_2. Adv Exp Med Biol. 2019. PMID: 31628650 Review.
Cited by
-
Combination of In Silico Methods in the Search for Potential CD4(+) and CD8(+) T Cell Epitopes in the Proteome of Leishmania braziliensis.Front Immunol. 2016 Aug 29;7:327. doi: 10.3389/fimmu.2016.00327. eCollection 2016. Front Immunol. 2016. PMID: 27621732 Free PMC article.
-
Emerging Concepts in Immune Thrombotic Thrombocytopenic Purpura.Front Immunol. 2021 Nov 11;12:757192. doi: 10.3389/fimmu.2021.757192. eCollection 2021. Front Immunol. 2021. PMID: 34858410 Free PMC article. Review.
-
Improved methods for predicting peptide binding affinity to MHC class II molecules.Immunology. 2018 Jul;154(3):394-406. doi: 10.1111/imm.12889. Epub 2018 Feb 6. Immunology. 2018. PMID: 29315598 Free PMC article.
-
Human CD4+ T Cells Specific for Merkel Cell Polyomavirus Localize to Merkel Cell Carcinomas and Target a Required Oncogenic Domain.Cancer Immunol Res. 2019 Oct;7(10):1727-1739. doi: 10.1158/2326-6066.CIR-19-0103. Epub 2019 Aug 12. Cancer Immunol Res. 2019. PMID: 31405946 Free PMC article.
-
New vistas unfold: Chicken MHC molecules reveal unexpected ways to present peptides to the immune system.Front Immunol. 2022 Jul 29;13:886672. doi: 10.3389/fimmu.2022.886672. eCollection 2022. Front Immunol. 2022. PMID: 35967451 Free PMC article. Review.
References
-
- Garcia KC, Degano M, Speir JA, Wilson IA. Emerging principles for T cell receptor recognition of antigen in cellular immunity. Rev Immunogenet (1999) 1:75–90 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials