

A heterozygous deletion mutation in the cardiac sodium channel gene SCN5A with loss- and gain-of-function characteristics manifests as isolated conduction disease, without signs of Brugada or long QT syndrome
- PMID: 23840796
- PMCID: PMC3695936
- DOI: 10.1371/journal.pone.0067963
A heterozygous deletion mutation in the cardiac sodium channel gene SCN5A with loss- and gain-of-function characteristics manifests as isolated conduction disease, without signs of Brugada or long QT syndrome
Abstract
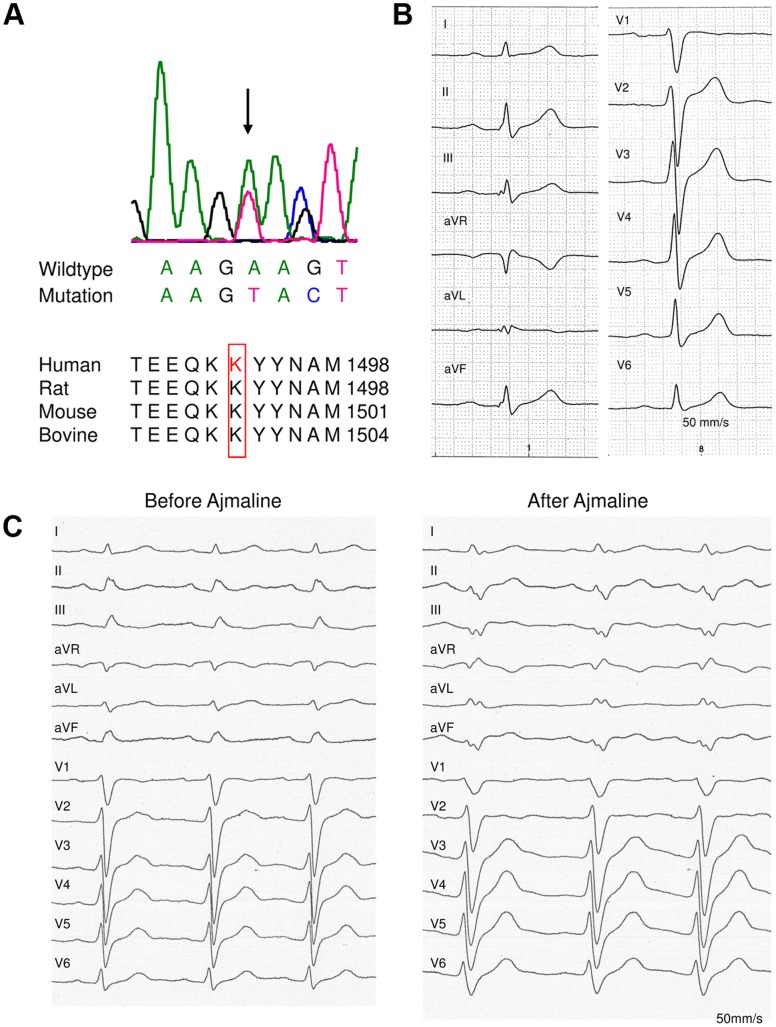
Background: The SCN5A gene encodes for the α-subunit of the cardiac sodium channel NaV1.5, which is responsible for the rapid upstroke of the cardiac action potential. Mutations in this gene may lead to multiple life-threatening disorders of cardiac rhythm or are linked to structural cardiac defects. Here, we characterized a large family with a mutation in SCN5A presenting with an atrioventricular conduction disease and absence of Brugada syndrome.
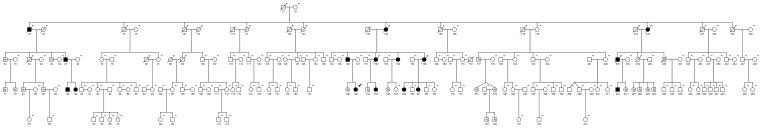
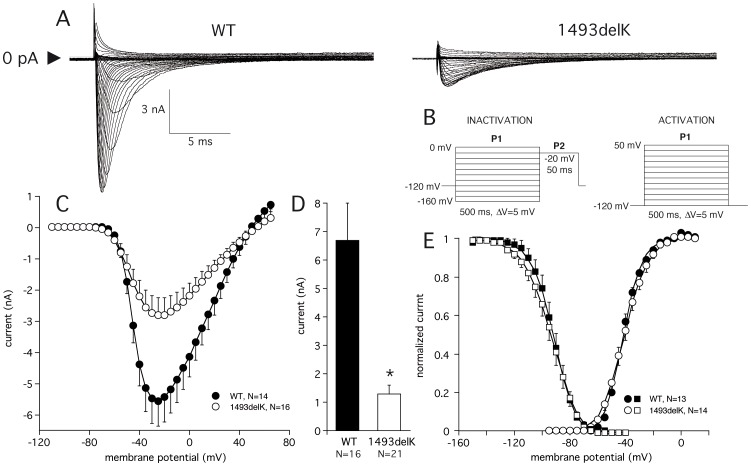
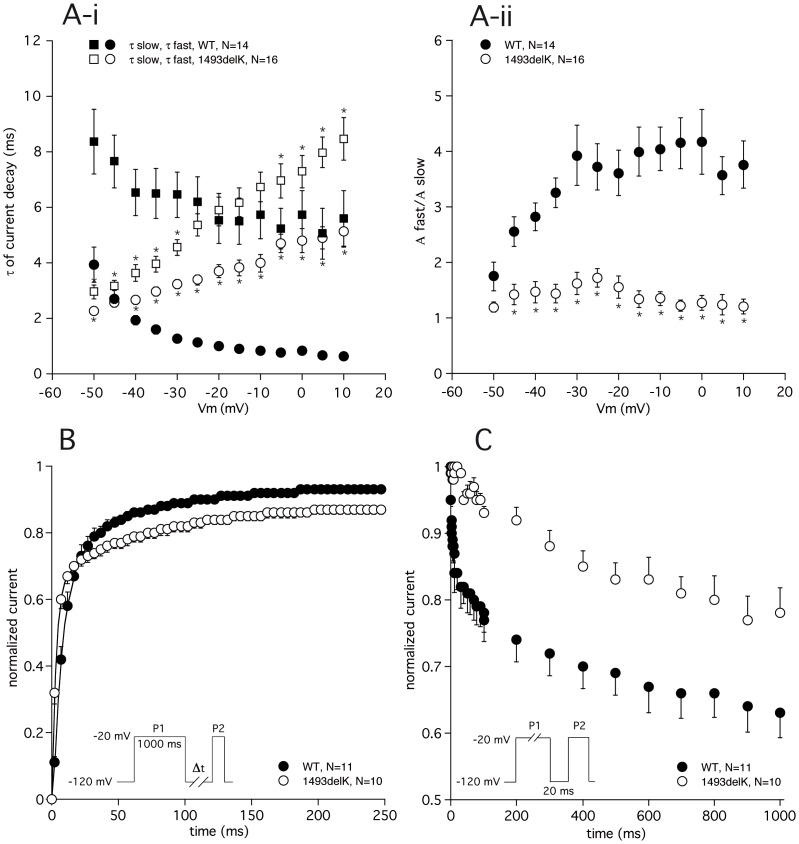
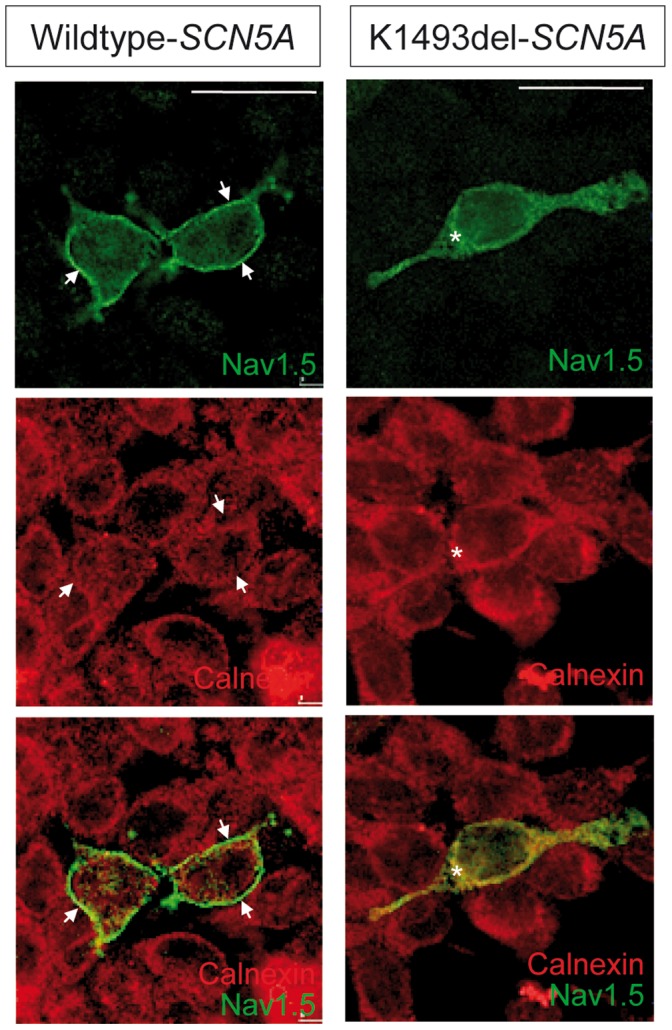
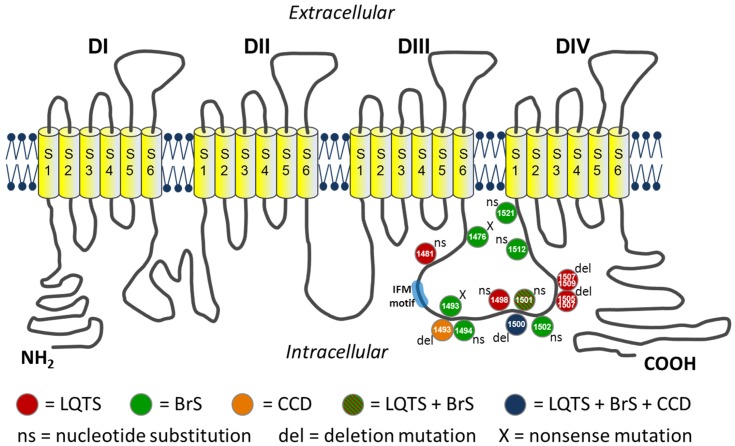
Method and results: In a large family with a high incidence of sudden cardiac deaths, a heterozygous SCN5A mutation (p.1493delK) with an autosomal dominant inheritance has been identified. Mutation carriers were devoid of any cardiac structural changes. Typical ECG findings were an increased P-wave duration, an AV-block I° and a prolonged QRS duration with an intraventricular conduction delay and no signs for Brugada syndrome. HEK293 cells transfected with 1493delK showed strongly (5-fold) reduced Na(+) currents with altered inactivation kinetics compared to wild-type channels. Immunocytochemical staining demonstrated strongly decreased expression of SCN5A 1493delK in the sarcolemma consistent with an intracellular trafficking defect and thereby a loss-of-function. In addition, SCN5A 1493delK channels that reached cell membrane showed gain-of-function aspects (slowing of the fast inactivation, reduction in the relative fraction of channels that fast inactivate, hastening of the recovery from inactivation).
Conclusion: In a large family, congregation of a heterozygous SCN5A gene mutation (p.1493delK) predisposes for conduction slowing without evidence for Brugada syndrome due to a predominantly trafficking defect that reduces Na(+) current and depolarization force.
Conflict of interest statement
Figures
Similar articles
-
A sodium channel pore mutation causing Brugada syndrome.Heart Rhythm. 2007 Jan;4(1):46-53. doi: 10.1016/j.hrthm.2006.09.031. Epub 2006 Sep 28. Heart Rhythm. 2007. PMID: 17198989 Free PMC article.
-
Novel SCN5A mutation leading either to isolated cardiac conduction defect or Brugada syndrome in a large French family.Circulation. 2001 Dec 18;104(25):3081-6. doi: 10.1161/hc5001.100834. Circulation. 2001. PMID: 11748104
-
Variable Na(v)1.5 protein expression from the wild-type allele correlates with the penetrance of cardiac conduction disease in the Scn5a(+/-) mouse model.PLoS One. 2010 Feb 19;5(2):e9298. doi: 10.1371/journal.pone.0009298. PLoS One. 2010. PMID: 20174578 Free PMC article.
-
[Cardiac sodium channelopathy from bench to bedside].Zhonghua Er Ke Za Zhi. 2013 Nov;51(11):874-7. Zhonghua Er Ke Za Zhi. 2013. PMID: 24484568 Review. Chinese. No abstract available.
-
Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy.JACC Clin Electrophysiol. 2018 May;4(5):569-579. doi: 10.1016/j.jacep.2018.03.006. Epub 2018 May 2. JACC Clin Electrophysiol. 2018. PMID: 29798782 Review.
Cited by
-
Congenital Long QT Syndrome: An Update and Present Perspective in Saudi Arabia.Front Pediatr. 2013 Nov 20;1:39. doi: 10.3389/fped.2013.00039. Front Pediatr. 2013. PMID: 24400285 Free PMC article. Review.
-
Relevance of mexiletine in the era of evolving antiarrhythmic therapy of ventricular arrhythmias.Clin Res Cardiol. 2024 Jun;113(6):791-800. doi: 10.1007/s00392-024-02383-9. Epub 2024 Feb 14. Clin Res Cardiol. 2024. PMID: 38353682 Free PMC article. Review.
-
Bringing home the bacon? The next step in cardiac sodium channelopathies.J Clin Invest. 2015 Jan;125(1):99-101. doi: 10.1172/JCI80014. Epub 2014 Dec 15. J Clin Invest. 2015. PMID: 25500878 Free PMC article.
-
MIR448 antagomir reduces arrhythmic risk after myocardial infarction by upregulating the cardiac sodium channel.JCI Insight. 2020 Dec 3;5(23):e140759. doi: 10.1172/jci.insight.140759. JCI Insight. 2020. PMID: 33108349 Free PMC article.
-
Conduction delays across the specialized conduction system of the heart: Revisiting atrioventricular node (AVN) and Purkinje-ventricular junction (PVJ) delays.Front Cardiovasc Med. 2023 Apr 19;10:1158480. doi: 10.3389/fcvm.2023.1158480. eCollection 2023. Front Cardiovasc Med. 2023. PMID: 37153461 Free PMC article.
References
-
- George AL Jr, Varkony TA, Drabkin HA, Han J, Knops JF, et al. (1995) Assignment of the human heart tetrodotoxin-resistant voltage-gated Na+ channel alpha-subunit gene (SCN5A) to band 3p21. Cytogenet Cell Genet 68: 67–70. - PubMed
-
- Wang Q, Li Z, Shen J, Keating MT (1996) Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics 34: 9–16. - PubMed
-
- Rook MB, Evers MM, Vos MA, Bierhuizen MF (2012) Biology of cardiac sodium channel Nav1.5 expression. Cardiovascular Research 93: 12–23. - PubMed
-
- Abriel H (2010) Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. Journal of Molecular and Cellular Cardiology 48: 2–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
